| Скачать .docx |
Дипломная работа: Основные закономерности сенсибилизированной фосфоресценции в твёрдых растворах органических соединений
ДИПЛОМНАЯ РАБОТА
ОСНОВНЫЕ ЗАКОНОМЕРНОСТИ СЕНСИБИЛИЗИРОВАННОЙ ФОСФОРЕСЦЕНЦИИ В ТВЁРДЫХ РАСТВОРАХ ОРГАНИЧЕСКИХ СОЕДИНЕНИЙ
Содержание.
Введение…………………………………………………………………………2
I. Основные закономерности сенсибилизированной фосфоресценции в твёрдых растворах органических соединений.
1.1. Особенности распределения примесных молекул в замороженных н.-парафиновых растворах при 77К………………………6
1.2. Особенности триплет-триплетного переноса энергии в н.-парафиновых растворах при 77К…………………………………………….9
1.3. Тушение люминесценции органических молекул в растворах различного рода ассоциатами………………………………………………16
II. Объекты исследования и техника эксперимента ……………...24
2.1. Объекты исследования…………………………………………..24
2.2. Техника эксперимента…………………………………………...31
III. Влияние отжига на параметры фосфоресценции дибромдифенилоксида и аценафтена в н.-октане ………………36
3.1.Обработка полученных данных…………………………………38
3.2.Основные результаты и выводы………………………………..44
Литература ………………………………………………………………….47
Введение.
С проблемой безызлучательного переноса энергии электронного возбуждения исследователям приходится сталкиваться при изучении самых разнообразных систем в таких областях науки как люминесценция, фотосинтез, радиационная физика и химия, биоэнергетика.
Фундаментальные представления о механизмах переноса энергии базируются в основном на классических результатах по фотонике синтетических органических соединений в конденсированных средах [1-3]. Хорошими модельными системами, которые часто используются для экспериментального изучения и проверки выводов теории переноса энергии триплетного возбуждения между молекулами, являются твёрдые растворы органических соединений. Это обусловлено своеобразием их физических свойств и возможностью широкого практического применения [4,5]. К таким средам относятся стекла активированные атомами или ионами, поликристаллические растворы, активированные полимерные пленки.
Основные закономерности межмолекулярного триплет-триплетного переноса энергии были установлены именно при исследовании тушения фосфоресценции молекул донора молекулами акцептора в этих системах. Однако даже для наиболее изученных донорно-акцепторных пар параметры переноса энергии триплетного возбуждения существенно отличаются у различных авторов [5-8].
Квантово – механическая теория триплет-триплетного переноса энергии в конденсированных средах была развита в работах Ферстера и Декстера [14,15].
Одним из выводов теории является то, что взаимодействие между компонентами донорно – акцепторной пары не влияет на константы скоростей как излучательной, так и безызлучательной дезактивации возбуждений акцептора. Именно это положение теории Фёрстера – Декстера (наряду с некоторыми другими) подвергается критике в новой теории переноса энергии, разрабатываемой в последнее время В.Я. Артюховым и Г.В. Майером [2]. Согласно этой теории взаимодействие между компонентами в донорно – акцепторной паре возмущает электронные состояния изолированных молекул еще до возбуждения молекул донора. При этом можно ожидать изменения константы скорости излучательной дезактивации энергии электронного возбуждения как в молекулах донора, так и в молекулах акцептора.
Наиболее актуальным вопрос о взаимном влиянии компонент донорно – акцепторной смеси на константы скоростей излучательной и безызлучательной дезактивации возбуждений является для межмолекулярного триплет – триплетного переноса энергии, поскольку он происходит при малых расстояниях между компонентами, так как обусловлен обменными взаимодействиями.
Таким образом, изучение механизмов дезактивации триплетных молекул в твердых растворах при их сенсибилизированном возбуждении и определение их вклада в дезактивацию возбуждений имеет актуальное значение для теории и практики межмолекулярного переноса энергии по обменно – резонансному механизму в конденсированных средах и является необходимым этапом дальнейшего развития его теоретических основ.
Актуальность работы. В настоящее время актуальным является исследование свойств органических люминофоров ввиду широкого применения их для решения задач как прикладного, так и фундаментального характера. Проведенные к настоящему времени исследования фосфоресценции твердых растворов, где возможна сенсибилизация энергии, показали, что концентрационное тушение можно уменьшить, использовав процесс отжига раствора. При этом наблюдается увеличение интенсивности фосфоресценции как донора, так и акцептора.Однако, как показали опыты проведенные Дерябиным М.И. и Куликовой О.И., увеличение интенсивности фосфоресценции молекул донора происходит в меньшее число раз. Данный эффект был обнаружен для смесей бензофенон-нафталин, бензофенон-аценафтен и антрон-флуорен в н.-парафинах. В моем дипломном проекте был заменен бензофенон в качестве донора энергии на дибромдифенилоксид. Если бензофенон, нафталин, аценафтен являются «звеньями одной цепи» (схожие по свойствам), то дибромдифенилоксид резко отличается от аценафтена по своим физико-химическим характеристикам и, поэтому, данное исследование представляет особый интерес.
Целью настоящей работы является исследование влияния отжига на параметры фосфоресценции донорно-акцепторной смеси дибромдифенилоксида и аценафтена в н.-октане. В соответствии с целью были поставлены следующие задачи :
1. Исследование изменения спектра фосфоресценции данной смеси под действием отжига.
2. Определение времени затухания фосфоресценции акцептора до и после отжига.
3. Изучение закона изменения интенсивности фосфоресценции акцептора от времени отжига.
4. Изучение закона зависимости константы скорости процесса от температуры (проверка аррениусовского характера процесса).
Дипломная работа состоит из введения, трех глав, заключения и списка литературы.
Первая глава носит обзорный характер. В ней приведены краткие сведения об основных направлениях исследования межмолекулярного триплет-триплетного переноса энергии в твердых средах. Также произведен анализ распределения примесных молекул в замороженных н.-парафинах. Дан обзор работ по концентрационному тушению триплетных состояний органических молекул в твердых растворах и влиянию температуры на эффективность переноса энергии триплетного возбуждения. Обосновывается постановка задач настоящей работы.
Во второй главе рассмотрены методические вопросы, где решаются проблемы выбора объектов исследования и подготовка техники эксперимента.
В третьей главе изложены результаты исследования влияния отжига образца на концентрацию триплетных молекул акцептора энергии. Установлено, что отжиг раствора может приводить как к увеличению интенсивности, так и к ее уменьшению (в зависимости от выбора используемых веществможет приводить как к увеличению интенсивности, так и к ее уменьшению ()_______________________________________________________).
Далее произведен анализ результатов работы. Рассмотрено влияние возможных механизмов концентрационного тушения в данных системах на параметры фосфоресценции донорно-акцепторной смеси и проведено сравнение их с экспериментальными данными. На основании этого сформулирован вывод о том, что отклонение результатов от полученных ранее связано с особенностью используемого донора энергии.
В заключении суммированы основные результаты и выводы.
Обнаруженное действие отжига как процесса, приводящего не только к снятию концентрационного тушения в н.-парафиновых растворах, но и к усилению тушения открывает перспективы дальнейшего исследования его применения к другим системам.
I . Основные закономерности сенсибилизированной фосфоресценции в твёрдых растворах органических соединений.
1.1. Особенности распределения примесных молекул в замороженных Н.-парафиновых растворах при 77К.
Молекулы растворенного вещества в н.-парафинах при кристаллизации могут оказаться внедренными в микрокристаллы парафина, либо могут быть вытеснены на поверхность. Молекулы примеси, внедренные в микрокристаллы, жестко фиксируются в них и хорошо изолированы друг от друга. Эти молекулы в случае слабого электрон-фононного взаимодействия дают квазилинейчатый спектр. Молекулы, адсорбированные на поверхности микрокристаллов, существуют либо в одиночном состоянии, либо в виде ассоциированных образований вплоть до агрегатов. Это зависит от концентрации примеси и «удобства» растворителя [60]. Для одиночных вытесненных молекул характерны сравнительно диффузные (хотя и достаточно структурные), полосатые молекулярные спектры, подобные таковым в стеклообразных растворителях. Ассоциация примесных молекул имеет преимущество в данном классе растворителей по сравнению со стеклообразными из-за создания их повышенных локальных концентраций при кристаллизации. В процессе роста микрокристаллов происходит вытеснение молекул примесей в оставшуюся область пространства. Агрегаты в ряде случаев характеризуются широкими бесструктурными полосами [60].
При высоких концентрациях в «удобных» растворителях часть молекул внедряется в кристаллы растворителя, а часть вытесняется. В неудобных же растворителях практически все молекулы вытесняются на поверхность. Поэтому локальная концентрация молекул примеси на поверхности кристаллов будет больше в неудобных растворителях, чем в удобных при одной и той же средней концентрации. Этим, по-видимому, объясняется то, что в неудобных растворителях аномальная температурная зависимость наблюдается при меньших концентрациях и более чётко выражена. Это даёт основание предположить, что процесс, ответственный за увеличение числа триплетных молекул, происходит на поверхности кристаллов. А так как сенсибилизированная фосфоресценция наблюдается в результате переноса энергии, то молекулы донора и акцептора, участвующие в процессе переноса находятся на поверхности кристаллов. Это подтверждает и вид спектров сенсибилизированной фосфоресценции для всех исследуемых соединений, который имеет диффузный характер.
В последние годы значительно возрос интерес к исследованиям взаимодействий между молекулами, адсорбируемыми на поверхности твёрдого тела [23-34]. Такие системы позволяют получить достаточно близкие расстояния между взаимодействующими молекулами, а так же интерес к таким системам обусловлен особенностями влияния микроскопической структуры матрицы на физические характеристики молекул.
Адсорбция примесей может быть получена на порошкообразных окисях магния и алюминия, на поверхности кремнозёма, в пористых и канальных матрицах. В качестве матриц используются стёкла, полученные по золь-гелевой технологии или путём выщелачивания натриевоборосиликатного стекла. Одна из особенностей заключается в том, что пространственное распределение молекул примесей носит фрактальный характер и характеризуется значительным разбросом расстояний между ближайшими соседними молекулами адсорбата, т.е. функция распределения молекул f ( r ) отличается от d - функции. Фракталы могут возникать либо в результате агрегации при диффузии (в них расстояние между ближайшими соседними частицами очень мало, постоянно и контролируется обычными, например ван-дер-ваальсовыми взаимодействиями между этими частицами), либо при взаимодействии с матрицей, вмещающей эти частицы [29].
В процессе отжига образца система из термодинамически неустойчивого состояния переходит в более устойчивое, которое соответствует более равномерному распределению молекул примеси. В результате чего часть молекул акцептора, которые ранее не участвовали в переносе энергии, попадают в сферу обменных взаимодействий с молекулами донора и теперь участвуют в излучении.
1.2. Особенности триплет-триплетного переноса энергии в Н.-парафиновых растворах при 77К.
Одним из распространенных механизмов дезактивации электронного возбуждения молекул является безызлучательный перенос энергии.
Безызлучательный перенос энергии электронного возбуждения представляет собой процесс, при котором возбуждённые молекулы донора энергии вступают во взаимодействие с невозбуждёнными молекулами акцептора энергии [10]. В результате такого взаимодействия появляется вероятность для перехода возбуждённой молекулы донора в электронно-колебательное состояние с меньшей энергией с одновременным переходом молекулы акцептора в состояние с большей энергией. В соответствии с законом сохранения энергии перенос энергии происходит только при условии, что спектры поглощения акцептора и спектры люминесценции донора перекрываются, т. е. в условиях резонанса.
Безызлучательный перенос энергии принято разделять на два вида: обменно-резонансный, когда перенос энергии осуществляется за счёт обменного взаимодействия, для возникновения которого необходимо перекрывание электронных облаков невозбуждённых молекул акцептора и возбуждённых молекул донора, и индуктивно-резонансный, когда электронные облака взаимодействующих молекул не перекрываются, а возбуждённые молекулы донора вступают в слабое кулоновское взаимодействие с невозбуждёнными молекулами акцептора.
Если электронные переходы в доноре и акцепторе разрешены правилами отбора, то перенос энергии происходит в результате диполь-дипольного взаимодействия. Для этого случая теория переноса энергии была развита Т. Фёрстером [14]. Она рассматривает процесс переноса энергии между молекулами в адиабатическом приближении и предполагает, что после переноса происходит быстрая колебательная релаксация в молекуле акцептора, что обеспечивает необратимость переноса энергии.
Вероятность переноса энергии в этом случае определяется из соотношения
![]() =
=  , (1.1)
, (1.1)
где ![]() - среднее время жизни возбужденного состояния донора в отсутствии тушителя,
- среднее время жизни возбужденного состояния донора в отсутствии тушителя, ![]() - расстояние между молекулами,
- расстояние между молекулами, ![]() - критическое расстояние переноса (фёрстеровский радиус) – расстояние, на котором вероятность переноса равна 1/
- критическое расстояние переноса (фёрстеровский радиус) – расстояние, на котором вероятность переноса равна 1/![]() . Величина
. Величина ![]() зависит от степени перекрывания спектров донора и акцептора, а так же пропорциональна силам осцилляторов переходов в доноре и акцепторе
зависит от степени перекрывания спектров донора и акцептора, а так же пропорциональна силам осцилляторов переходов в доноре и акцепторе
![]() ~
~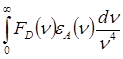 , (1.2)
, (1.2)
где ![]() - волновое число, FD
(
n
) -
квантовый спектр излучения донора, e
А
(
n
)
- спектр поглощения акцептора; оба спектра нормированы на единичную площадь.
- волновое число, FD
(
n
) -
квантовый спектр излучения донора, e
А
(
n
)
- спектр поглощения акцептора; оба спектра нормированы на единичную площадь.
Развитая Фёрстером теория явилась той основой, на которой базировалось дальнейшее изучение переноса энергии в случае обменных взаимодействий. Перенос энергии при обменном взаимодействии наблюдается, когда электронные переходы в акцепторе запрещены. В работе [15] Декстер Д. Л. показал, что в отличие от всех видов кулоновских взаимодействий, при обменных взаимодействиях константа переноса не зависит от силы осцилляторов переходов в доноре и акцепторе энергии, а зависимость её от расстояния в паре имеет экспоненциальный характер:
![]() ~
~ , (1.3)
, (1.3)
где L - средний эффективный боровский радиус возбуждённой молекулы донора и невозбуждённой акцептора.
Экспоненциальный множитель появляется вследствие того, что электронная плотность в молекуле, начиная с некоторой точки, спадает с расстоянием по экспоненте. Таким образом, при обменно-резонансных взаимодействиях вероятность переноса энергии уменьшается с увеличением расстояния между молекулами гораздо быстрее, чем в случае индуктивно-резонансных взаимодействий.
Межмолекулярный триплет-триплетный перенос энергии электронного возбуждения был впервые обнаружен в 1952г. Терениным А.Н. и Ермолаевым В.Л. в твердых растворах органических соединений [9]. Они наблюдали новое явление, заключающееся в том, что фосфоресценция нафталина в твердом растворе возбуждалась светом ртутной лампы с длинной волны в области 365 нм в присутствии бензофенона или бензальдегида в растворе хотя сам нафталин излучение с данной длинной волны не поглощает.
Основные закономерности триплет-триплетного переноса энергии между молекулами были установлены В.Л Ермолаевым при изучении данного явления для органических соединений в твердых растворах. Эти закономерности были выявлены на основании изучения влияния акцептора на параметры фосфоресценции донора и особенностей сенсибилизированной фосфоресценции.
При экспериментальном изучении явления сенсибилизированной фосфоресценции донорно-акцепторные пары обычно выбирают таким образом, чтобы они удовлетворяли трем ниже перечисленным условиям:
1) триплетный уровень молекул акцептора расположен также или ниже соответствующего уровня молекул донора (закон сохранения энергии).
2) Первый возбужденный синглетный уровень молекул акцептора был выше соответствующего уровня молекул донора. Это позволяет возбуждать донор энергии не затрагивая при этом молекулы акцептора.
3) Время жизни триплетных молекул донора намного меньше времени жизни триплетных молекул акцептора. Выполнение этого условия позволяет отделить во времени фосфоресценцию молекул акцептора от фосфоресценции донора.
Если выполнение первого условия является необходимым для осуществления триплет-триплетного переноса энергии, то выполнение последних двух необязательно. Они необходимы лишь для удобства эксперимента.
В.Л. Ермолаев и А.Н. Теренин показали, что в спектрах поглощения донорно-акцепторной смеси, отсутствуют какие-либо новые полосы по сравнению с суммой спектров компонентов [80]. Спектры сенсибилизированной фосфоресценции акцепторов тождественны спектрам их фосфоресценции, возбуждаемых прямо в их полосу синглет-синглетного поглощения. Эти результаты однозначно показали, что возбуждение молекул акцептора не связано со смещением их триплетного уровня. Однако, они не могут дать однозначного ответа на вопрос влияет ли взаимодействие между компонентами донорно-акцепторной пары на положение триплетного уровня акцептора или нет. Это связано с тем, что спектры сенсибилизированной фосфоресценции акцептора широкие и на их параметры (ширину полос, положение максимума 0-0 полосы, ее форму и др.) существенно влияет как неоднородное взаимодействие молекул акцептора с раствором, так и молекул акцептора между собой. Поэтому небольшие изменения положения триплетного уровня могут маскироваться другими явлениями (например концентрационным смещением и уширением спектра и т. д.). Следовательно, для ответа на вопрос на сколько сказывается возмущение молекулами донора соответствующего электронного состояния акцептора на положение триплетного уровня последнего, необходимо создать условия, при которых смещение триплетного уровня молекул акцептора в пределах неоднородно уширенной полосы, обусловленное взаимодействиями в донорно-акцепторной паре, можно выделить и исследовать.
В кристаллах физическая картина переноса энергии существенно отличается от других систем. Из-за трансляционной симметрии возможно возбуждение любой элементарной ячейки кристалла или же любой составляющих кристалл молекул. В этом случае перенос энергии обусловлен движением квазичастиц - экситонов. Экситонная теория переноса энергии в кристаллах заняла, судя по широкому кругу рассматриваемых интересов, самостоятельную область в разделе межмолекулярных взаимодействий.
В жидких растворах и газах перенос энергии электронного возбуждения контролируется диффузией. Диффузионные процессы также приводят к увеличению вероятности процессов, приводящих к безызлучательной дезактивации электронного возбуждения, за счёт чего фосфоресценция в жидкости затухает намного быстрее, чем в твёрдых растворах.
В твёрдых растворах молекулы находятся в триплетном состоянии более длительное время, поэтому представляется удобным исследовать основные характеристики межмолекулярного переноса энергии именно в данных системах. Исследования в данном направлении можно разделить по типу центров, между которыми наблюдается перенос энергии: а) между одиночными молекулами различных примесей, б) между одинаковыми молекулами примесей (миграция энергии), либо в) от основы (матрицы растворителя) к молекулам примеси.
В стеклообразных растворах при 77 К спектры фосфоресценции акцептора имеют диффузный характер как при прямом, в отсутствие донора, так и при сенсибилизированном возбуждении и заметного различия между ними не наблюдается. Поэтому извлечь какую – либо информацию об особенностях взаимодействия партнеров в донорно – акцепторной паре из спектров фосфоресценции достаточно сложно. По видимому, это и является причиной того, что их изучению посвящено сравнительно малое число работ, имеющихся в литературе.
Новые возможности для спектральных исследований переноса энергии дает открытый в 1952 г. Э.В. Шпольским, А.А. Ильиной и Л.А. Климовой эффект резкого сужения спектральных полос люминесценции ряда ароматических углеводородов в замороженных н.- парафиновых растворах [60]. Попытки получить квазилинейчатый спектр [72-76] сенсибилизированной фосфоресценции не дали положительного результата. Тонкая структура спектра излучения акцептора размывалась при переходе к сенсибилизированному возбуждению. Квазилинейчатые спектры сенсибилизированной фосфоресценции удавалось получить лишь в том растворителе, в котором и акцептор и донор имеют каждый в отдельности при выбранной концентрации квазилинейчатые спектры [35-38]. Было установлено, что эффективность образования донорно – акцепторных пар в этих условиях различна для различных центров. Это проявляется в отличии мультиплетной структуры спектров при прямом, в отсутствие донора, и сенсибилизированном возбуждении, что объясняется образованием нескольких излучающих и поглощающих центров с разной эффективностью передачи энергии. Причина различной эффективности переноса энергии связывается с зависимостью обменно – резонансного взаимодействия от взаимной ориентации партнеров в матрице растворителя. Так же были изучены спектры сенсибилизированной фосфоресценции хинолина и нафталина в матрицах н.- парафинов от пентана до октана при 77 К [77]. Из сопоставления мультиплетов обычной и сенсибилизированной фосфоресценции сделан вывод, что они различаются как по числу компонентов, так и по положению и относительной интенсивности. Было выдвинуто предположение, что мультиплетность в спектре акцептора при сенсибилизированном возбуждении и его квазилинейчатая структура обусловлены эффектом селекции в переносе энергии. Этот эффект селекции может быть связан как с особенностями взаимного расположения энергетических уровней донора и акцептора, так и с особенностями взаимного расположения партнеров в донорно – акцепторной паре. Эту гипотезу авторы [77] подтверждают различием мультиплетной структуры спектров сенсибилизированной фосфоресценции акцептора в одном и том же растворителе в случае различных доноров. Однако возможна и иная интерпретация результатов этой работы. Не исключено, что за квазилинейчатые спектры, ответственны молекулы акцептора, находящиеся в агрегатах донора. Так в некоторых работах [78,79] наблюдался квазилинейчатый спектр сенсибилизированной фосфоресценции нафталина в кристаллах бензофенона при возбуждении через основу. И было установлено, что триплет – триплетный перенос энергии эффективно осуществляется, если молекулы акцептора внедрены в агрегаты донора.
Следует отметить, что даже для наиболее структурных спектров квазилинии сенсибилизированной фосфоресценции уширены в сравнении с квазилиниями обычной фосфоресценции в тех же условиях [35,36]. Связано ли это уширение только с влиянием донора на формирование микроматрицы или же здесь проявляется непосредственное влияние донора на параметры фосфоресценции акцептора – дать однозначный ответ на этот вопрос, на основании экспериментального материала имеющегося к настоящему времени, не представляется возможным.
1.3. Тушение люминесценции органических молекул в растворах различного рода ассоциатами.
В обзоре Южакова В.И. [29], обобщающем результаты экспериментальных и теоретических работ по концентрационному тушению люминесценции, показано, что к тому времени наметились два основных подхода в объяснении ее природы. Первый основывался на возможности индукционной резонансной миграции электронного возбуждения между мономерными молекулами красителя. Эти представления затем были заложены в основу миграционной теории концентрационного тушения люминесценции. Согласно данной теории, тушение при больших концентрациях люминесцирующих веществ происходит за счет резонансной передачи энергии электронного возбуждения от одной молекулы красителя, находящейся в мономерной форме к другой такой же молекуле. При этом часть таких переходов сопровождается тушением.
Другой подход в объяснении концентрационного тушения люминесценции подчеркивал важность обратимой ассоциации молекул люминесцирующих веществ. Это явление объяснялось неактивным поглощением нелюминесцирующих ассоциатов. Ассоциационная теория концентрационного тушения люминесценции, созданная Левшиным В.Л., предусматривает собственное неактивное поглощение ассоциатов и миграцию возбуждения с мономеров на эти ассоциаты.
В дальнейшем миграционная теория концентрационного тушения люминесценции была развита в ряде теоретических работ [3, 4, 30-37] и подтверждена экспериментально [21,22,37-42].
Под миграцией энергии подразумевается передача возбуждения только между центрами одинаковой природы. В зависимости от природы возбуждений их перенос осуществляется либо дальнодействующим (мультипольным), либо короткодействующим (обменным) межцентровым взаимодействием.
Делокализация возбуждения по системе случайно расположенных одинаковых центров складывается в диффузию. В работах [5,8,45] методами теории неупорядоченных систем найдена концентрационная зависимость коэффициента диффузии как при мультипольном, так и при обменном взаимодействии.
Однако возможность диффузии возбуждения на большие расстояния вовсе не означает, что его тушение обязательно является диффузионным. Зона тушения вокруг акцептора может быть настолько узка, что возбуждение способно попасть внутрь неё и выйти наружу однократным перемещением, а не последовательностью мелких шагов, складывающихся в континуальную диффузию. Одноактное тушение называют прыжковым. Скорости диффузионного и прыжкового тушения по разному зависят от концентрации доноров и микропараметров переноса возбуждения [5,8]. В разбавленных растворах, по мнению авторов [45], следует отдать предпочтение прыжковому механизму тушения.
В обзорах Бодунова Е. Н. [7,8] проведён анализ различных теоретических методов: Монте-Карло, непрерывных во времени случайных блужданий, эффективной среды и самосогласованный графический, используемых при исследовании спектральной миграции возбуждения в трёхмерных средах. Анализируется зависимость положения и формы неоднородно уширенного спектра люминесценции от времени и концентрации молекул при различных условиях возбуждения среды и механизмах межчастичного взаимодействия. Приводятся концентрационные зависимости квантового выхода люминесценции сред, содержащих два сорта молекул (доноров и акцепторов энергии возбуждений).
При вычислении параметров люминесценции в [7,8] основное внимание уделяется мультипольному взаимодействию. Для обменного взаимодействия вычисляется лишь коэффициент диффузии возбуждения. Это по-видимому связано с тем, что обменные взаимодействия осуществляются на меньших расстояниях по сравнению с мультипольными. Если индуктивно-резонансные взаимодействия разрешены правилами отбора, то они обладают преимуществом перед обменно-резонансными. Но если передача энергии по всем видам кулоновского взаимодействия запрещена, как в случае передачи энергии триплетного возбуждения, то обменный механизм миграции возбуждения является основным. Недостаток теоретических работ, рассматривающих влияние миграции триплетного возбуждения по системе случайно расположенных центров на параметры выхода их фосфоресценции, делает сложным выявление данного механизма тушения в рассматриваемых системах.
В работе [49] показано, что миграция энергии возбуждения в условиях неоднородного уширения спектров растворённого вещества при определённом соотношении между временами жизни возбуждённого состояния, миграции и релаксации приводит к концентрационному длинноволновому смещению спектров люминесценции органических красителей в различных растворителях. Как в твёрдых телах, так и в жидких растворах центры люминесценции одних и тех же веществ не являются идентичными вследствие различия их ближайшего окружения. При этом, помимо смещения энергетических уровней примесных центров, от величины локальных полей зависят и вероятности излучательных и безызлучательных переходов в молекулах растворенного вещества, а следовательно, и времена жизни возбуждённого состояния [29]. Направленность миграционных процессов при наличии расстройки энергетических уровней взаимодействующих молекул с повышением концентрации растворённого вещества приводит к увеличению заселённости первого возбуждённого состояния молекул с наиболее низко расположенными уровнями энергии. Направленная миграция на такие молекулы проявляется в концентрационном длинноволновом смещении спектров свечения. Если величина квантового выхода люминесценции молекул значительно уменьшается с понижением их возбуждённых уровней, то при этом также возникает концентрационное тушение люминесценции.
Авторами [39] показано, что механизм миграции по мономерным молекулам обнаруживается только при отсутствии в растворе ассоциатов.
Увеличение концентрации раствора обычно сопровождается развитием межмолекулярных взаимодействий, часто приводящих к ассоциациимолекул различной степени сложности. В результате в растворе наряду с мономерными молекулами появляются центры, существенно изменяющие оптические свойства раствора. Экспериментально наблюдаются разнообразные изменения спектров поглощения и люминесценции растворов, падение квантового выхода свечения и других параметров [44-48]. Это связано со сложными межмолекулярными взаимодействиями в растворах органических соединений и различной природой сил, объединяющих молекулы и ассоциаты.
Образование ассоциатов может происходить как за счет сил Ван-дер-Ваальса, так и благодаря возникновению водородных связей [29].
Среди органических молекул наиболее изученными с точки зрения образования ассоциатов являются молекулы красителей и класса хлорофиллов [29,43]. Очень хорошо молекулы красителей ассоциируют в воде, в смесях полярных и неполярных растворителей.
Образование ассоциатов может происходить и между различными молекулами. Разнородные ассоциаты обладают спектральными свойствами, отличными от мономеров и однородных ассоциатов [50]. Они могут влиять на характер процессов переноса энергии возбуждения в смешанных растворах и служить дополнительными центрами ее захвата.
Авторами [50] обнаружено существование как люминесцирующих разнородных ассоциатов – родамин 6Ж + метиленовый голубой, так и нелюминесцирующих – родамин 6Ж + бензопурпурин 4Б в буферных водных растворах.
Как упоминалось выше, концентрационное тушение люминесценции вследствие неоднородного уширения спектров может быть обнаружено только при отсутствии в растворе более активных центров тушения. Таковыми являются либо другие примеси, эффективно осуществляющие тушение, либо ассоциированные образования. В работе [50] показано, что перенос энергии на разнородные ассоциаты осуществляется с большей скоростью, чем на однородные или на мономеры.
Процесс образования ассоциатов может осуществляться как между разнородными молекулами примесей, так и между молекулами примесей и растворителя. Например, в работе [51] исследованы молекулы аминокумаринов, которые обладают протонно-акцепторными свойствами. Взаимодействуя с молекулами протонно-донорного растворителя – этанола, они образуют комплексы с водородной связью. Н-связь возникает между карбонильной группой (>С=О) молекул красителей и оксигруппой (-ОН) растворителя. Вычислена энергия связи – 1400см-1 . Таким образом, ассоциация молекул приводит к самым разным изменениям в спектрах поглощения и люминесценции молекул примесей. Это говорит о том, что вопрос о природе сил, объединяющих молекулы в ассоциаты, должен решаться в каждом конкретном случае по-своему. Силы же Ван-дер-ваальса, как универсальные силы, всегда действуют между молекулами на достаточно близком расстоянии, однако в некоторых случаях теряют первостепенную роль.
В жидкости процесс ассоциации является обратимым. Все концентрационные эффекты полностью исчезают, а первоначальные оптические свойства растворов, содержащих мономерные молекулы исследуемого вещества, полностью восстанавливаются при обратном разведении концентрированных растворов [29].
Понижение температуры растворов сдвигает равновесие между мономерными молекулами и ассоциированными в сторону ассоциатов [29].
Кроме ассоциатов молекул в основном состоянии, возможно образование возбужденных димеров. Они состоят из возбужденной и невозбужденной молекул, объединяющихся за время меньшее, чем средняя длительность их возбужденного состояния. Эти возбужденные димеры, получившие название эксимеров (одинаковые молекулы), эксиплексов (разные молекулы), обуславливают сильные изменения спектра люминесценции при постоянстве спектра поглощения [52-54].
Подводя итог анализу литературных данных по процессу ассоциации органических молекул, можно сделать вывод, что основными объектами при его исследовании являлись жидкие растворы молекул красителей. Процесс ассоциации ароматических углеводородов в твердых растворах остается малоизученным, тогда как именно на этих системах проверены основные параметры межмолекулярного триплет-триплетного переноса энергии.
Обобщив наметившиеся к настоящему времени подходы к вопросу концентрационного тушения возбужденных состояний можно утверждать, что его причинами являются либо физико-химические (ФХ), либо резонансные (Р) взаимодействия между молекулами. Причём последние могут иметь обменный (близкодействующий), либо мультипольный (дальнодействующий) характер.
Эти причины могут приводить к изменению спектров поглощения и люминесценции, падению квантового выхода свечения, уменьшению времени его затухания. Основные механизмы, обуславливающие изменение параметров свечения в результате данных взаимодействий следующие:
1. Неактивное поглощение света ассоциатами (ФХ взаимодействие);
2. Миграция энергии между мономерными молекулами по индуктивно-резонансному или по обменно-резонансному механизму (Р взаимодействие);
3. Миграция энергии на ассоциаты (ФХ и Р взаимодействие);
4. Изменение внутримолекулярных безызлучательных констант. (ФХ и Р взаимодействие).
В литературе последний механизм тушения изучен недостаточно подробно, однако имеются факты, подтверждающие его существование и в ряде случаев его первостепенное значение. К одному из них можно отнести вопрос о том, почему образовавшиеся ассоциаты в основном не люминесцируют или слабо люминесцируют [29].
Рассмотренные механизмы могут обуславливать также тушение возбужденных состояний в условиях переноса энергии. Под таким углом зрения этот вопрос до настоящего времени не рассматривался. Хотя очевидно, что благоприятные для тушения условия создаются именно в концентрированных растворах, которые и являются необходимыми для наблюдения переноса энергии, особенно по обменно-резонансному механизму. Расхождения в определении параметров переноса энергии между органическими молекулами в твердых растворах ранее не связывались ни с возможным тушением, вызванным миграцией энергии, ни с влиянием ассоциатов на процесс перераспределения энергии в системе. Процесс ассоциации ароматических углеводородов так же остаётся малоизученным, тогда как именно на этих системах проверены основные параметры межмолекулярного триплет-триплетного переноса энергии.
Концентрационное поведение спектров излучения и поглощения однородных примесей ароматических углеводородов в н.-парафиновых твердых растворителях исследовалось многими авторами (например, [55-59]).
Шпольским Э.В. с сотрудниками [55] были исследованы спектры флуоресценции и поглощения нафталина в н.-гептане и циклогексане при 77К в диапазоне концентраций 10-2 – 10-4 М. Спектры флуоресценции растворов нафталина, состоящие при малых концентрациях из сравнительно широких полос, приобретают при увеличении концентрации квазилинейчатый характер. Авторы объясняют это следующим образом: «…за квазилинейчатые спектры флуоресценции и поглощения ответственны изолированные молекулы вещества «устроившиеся» в кристаллической матрице растворителя. Оказывается, что число таких молекул ограничено, так что для разных соединений существует предельная концентрация, выше которой интенсивность квазилинейчатого спектра не возрастает; если концентрация несколько превышает предельную, то на квазилинейчатый спектр флуоресценции накладывается полосатый молекулярный спектр, аналогичный спектру данного соединения в стеклообразном растворителе. Таким образом, за спектр флуоресценции растворов нафталина малых концентраций ответственны молекулы, «неустроенные» в кристаллической матрице растворителя. Повышение концентрации раствора приводит к агрегации таких молекул, чему способствует кристаллический характер матрицы. Агрегаты нафталина характеризуются своим собственным спектром поглощения и не люминесцируют». Болотниковой Т.Н. и Наумовой Т.М. [56] установлено аналогичное поведение спектров фосфоресценции замороженных растворов нафталина в гексане и фенантрена в октане при изменении концентраций от 10-5 до 10-1 М.
Таким образом, в н.-парафиновых растворах концентрационное тушение люминесценции наблюдается при более низких средних концентрациях, чем в стеклообразных матрицах. Это достигается созданием повышенных локальных концентраций молекул примесей на поверхностях микрокристаллов растворителя при вытеснении «лишних» молекул, превышающих предельную концентрацию «устроенных». Так как предел концентрации «устроенных» молекул определяется «удобством» растворителя, то в «неудобных» растворителях практически все молекулы примесей будут находиться на поверхности микрокристаллов. В таких условиях даже при низких концентрациях молекул примесей могутвозникать условия, способствующие концентрационному тушению люминесценции, так же как и переносу энергии. Исследованию механизмов тушения люминесценции в «неудобных» н.-парафиновых растворителях в литературе уделено недостаточное внимание. Тем не менее, на наш взгляд, они являются хорошими модельными системами, позволяющими изучать особенности переноса энергии в условиях концентрационного тушения люминесценции.
II . Объекты исследования и техника эксперимента.
2.1. Объекты исследования.
Важнейшим источником информации о строении и свойствах молекул и твердых тел являются их оптические спектры [81-83]. Для решения поставленных задач особый интерес представляют электронные спектры, поскольку именно в них наиболее отчетливо проявляется связь оптических свойств молекулы (или кристалла) с химическими, фотофизическими и фотохимическими свойствами. Но наиболее важным для нас является то, что электронные спектры оказываются наиболее чувствительными к различного рода внутри- и межмолекулярным взаимодействиям и служат ценным средством исследования взаимодействия молекул между собой и с окружением [84-87]. Поэтому метод оптической спектроскопии был выбран в качестве одного из основных методов исследования.
В экспериментальных исследованиях триплетных молекул важное место, наряду со спектральными, занимают кинетические методы [1,2], то есть изучение процессов заселения и распада возбужденных состояний. Определенные из кинетических экспериментов параметры являются характеристиками, как самих молекул, так и их взаимодействия между собой и с матрицей, в случае примесных центров. Особенно важным является то, что параметры кинетики (время накопления и время дезактивации возбужденных состояний), определяются константами скоростей соответствующих переходов и, следовательно, позволяют извлечь информацию, о путях дезактивации триплетно возбужденных молекул. Этим обусловлена необходимость использования кинетических методов для установления и изучения механизмов дезактивации триплетных состояний органических молекул в твердых матрицах при их сенсибилизированном возбуждении.
В начале проведения эксперимента необходимо было определиться с объектами исследования, а именно с выбором веществ (молекулы донорно-акцепторной пары), которые должны удовлетворять следующим требованиям [16]:
1. Триплетный уровень молекул донора энергии должен быть расположен выше триплетного уровня молекул акцептора (закон сохранения энергии).
2. Для осуществления избирательного возбуждения только молекул донора энергии их флуоресцентный уровень должен быть ниже соответствующего уровня молекул акцептора.
При выполнении этих условий синглет-синглетный перенос энергии невозможен из-за неблагоприятного расположения энергетических уровней, а триплет-триплетный перенос наблюдается, если молекулы находятся в радиусе обменных взаимодействий.
Время затухания всех молекул акцепторов в замороженных растворах при 77 К составляет несколько секунд, что на два порядка больше времени затухания фосфоресценции доноров. Благодаря этому, после прекращения возбуждения уже спустя 0,1 секунды свечение полностью определяется фосфоресценцией акцептора.
Из этих соображений в качестве донора был взят дибромдифенилоксид, а так же для сравнения ранее изученный бензофенон, квантовый выход триплетных состояний которого близок к единице [62]. В качестве акцептора – аценафтен.
Аценафтен . Спектры флуоресценции и фосфоресценции аценафтена в н.-парафинах изучены достаточно подробно при различных концентрациях примеси Мамедовым Х. И. [68] и DekkersJ. J. [69]. Как и для остальных рассматриваемых соединений вид спектра люминесценции аценафтена зависит от подбора растворителя и концентрации примеси.
Наиболее удобным растворителем для аценафтена является н.-пентан, в котором спектр люминесценции в широком диапазоне конценцтраций (10-5 -10-2 М) является квазилинейчатым [69]. При дальнейшем увеличении длины цепочки растворителя для малых концентраций спектр преобразуется в диффузные полосы. Так, в н.-гексане такой предельной концентрацией является 10-5 М , в н.-гептане –10-4 М, а в н.-октане квазилиний не наблюдается вообще. Представляет интерес уменьшение интенсивности свечения в последнем растворителе приблизительно на порядок при увеличении концентрации от 10-4 М до 10-2 М. Максимум диффузной полосы при этом немного смещается в коротковолновую область, а в длинноволновой области, отстоящей более чем на 1000 см-1 , появляется широкое диффузное свечение, принадлежащее кристаллическому аценафтену.
При исследовании концентрационной зависимости спектров фосфоресценции аценафтена в матрицах н.-гексана при 77 К [70] наблюдалось три типа молекулярных спектров. Для концентраций раствора от 10-2 М до10-4 М наблюдался квазилинейчатый спектр. Для концентраций раствора, меньших чем 10-3 М, в спектре с коротковолновой стороны от квазилиний наблюдались широкие молекулярные полосы, смещенные на 50 см-1 и подобные полосам в спектре флуоресценции при тех же концентрациях. В узком интервале концентраций в области 10-1 М наряду с квазилиниями появились полосы, смещенные в длинноволновую область спектра относительно квазилиний на 200 см-1 . На основании результатов температурной зависимости спектров фосфоресценции аценафтена авторами выдвинуто предположение, что за первый тип центров отвечают молекулы, внедренные в кристаллы, за второй – одиночные молекулы аценафтена, вытесненные на поверхность. Третий тип обусловлен свечением центров, внедренных в кристаллы н.-гексана, однако большая ширина и их смещение, по-видимому, связаны с неоднородным уширением и увеличением электрон-фононного взаимодействия из-за высоких концентраций.
В работе [13] был исследован спектр и кинетика сенсибилизированной фосфоресценции аценафтена в кристаллическом бензофеноне при переносе энергии от основы к примеси. Работа была выполнена с целью исключить из рассмотрения данный тип центров (микрокристаллы донора с внедренными в них молекулами акцептора) при условиях создания больших концентраций примеси (10-1 М). Как показал проведенный анализ, максимум 0-0 полосы сенсибилизированной фосфоресценции смещен на 120 см-1 в длинноволновую область по отношению к максимуму этой же полосы в н.-гексане. Полуширина 0-0 полосы сенсибилизированной фосфоресценции аценафтена в кристаллическом бензофеноне составляет около 240 см-1 . Среднее значение времени затухания для интегральной ( без разложения в спектр) интенсивности составляет 2.40 с, что заметно отличается от среднего времени затухания сенсибилизированной фосфоресценции аценафтена в н.-гексане.
Достоверные тонкоструктурные спектры аценафтена в основном и возбужденном электронных состояниях, не искаженные влиянием на молекулы окружающей среды, получены в [71] при охлаждении в сверхзвуковой струе.
Растворители Н.-парафиновые растворители при быстром замораживании кристаллизуются и представляют собой поликристаллическую снегообразную массу. В процессе кристаллизации молекулы активатора могут внедряться в кристаллики растворителя, либо вытесняться в свободное пространство между ними или различного рода дефекты [61]. В «неудобных» растворителях молекулы примеси вытесняются из кристаллов практически полностью. Поэтому локальная концентрация примеси может быть намного больше, чем средняя концентрация ее в стеклообразных растворах при тех же условиях. И это дает нам возможность исследовать особенности переноса энергии при меньших расстояниях между молекулами в донорно-акцепторной паре, а также становится более доступным исследование механизмов концентрационного тушения триплетных состояний. По-видимому, в замороженных н.-парафиновых растворах донорно-акцепторных смесей существует два процесса, влияющих на концентрацию триплетных молекул акцептора энергии, а следовательно и на интенсивность сенсибилизированной фосфоресценции.
Первый процесс – тушение триплетных состояний молекул донора акцептором энергии, приводит к падению интенсивности сенсибилизированной фосфоресценции при повышении температуры и сопровождается падением t . Падение интенсивности сенсибилизированной фосфоресценции в результате этого процесса происходит во всём исследованном интервале температур, в том числе и в аномальной области 2. Этот процесс практически не зависит от концентрации молекул примеси в растворе.
Второй процесс ведет к увеличению числа триплетных молекул. Прирост числа триплетных молекул акцептора энергии в результате этого процесса зависит как от концентрации раствора, так и от температуры.
Исходя из всего этого в качестве растворителя для решения поставленной задачи был выбран н.-октан, точка плавления которого 216К.
2.2. Техника эксперимента.
Основными экспериментами для решения поставленной задачи являлись изучение кинетики накопления и распада триплетных молекул акцептора и донора энергии, изучение спектров обычной и сенсибилизированной фосфоресценции, а также влияние на них температуры.
Экспериментальные исследования были выполнены на установке на базе спектрометра ДФС-12 с дифракционной решёткой 600 штр./мм и линейной дисперсией 5 ![]() /мм, блок схема которой приведена на рис. 2.1. Она позволяла получать и исследовать спектры поглощения и люминесценции, кривые разгорания и затухания фосфоресценции, а также зависимости люминесцентных характеристик изучаемых объектов от температуры.
/мм, блок схема которой приведена на рис. 2.1. Она позволяла получать и исследовать спектры поглощения и люминесценции, кривые разгорания и затухания фосфоресценции, а также зависимости люминесцентных характеристик изучаемых объектов от температуры.
Изучаемый раствор необходимой концентрации помещался в цилиндрическую кварцевую кювету. Исследуемый раствор охлаждался путём быстрого погружения в кварцевый прозрачный сосуд Дьюара с кипящим азотом. Такой способ охлаждения растворов можно назвать быстрым замораживанием в отличие от «медленного» замораживания, которое осуществлялось в парах азота. В последнем случае кристаллизация раствора происходила за 5-10 мин.
Молекулы донора энергии возбуждались светом ртутной лампы ПРК-2 с фильтром 365 нм. Молекулы, используемые в качестве акцепторов энергии, излучение с данной длиной волны не поглощают. При исследовании обычной фосфоресценции молекул акцептора (аценафтен), последние возбуждались излучением ПРК-2 с фильтром 313 нм.
Градуировка спектрометра производилась по линиям излучения ртутной лампы низкого давления. Ширина входной и выходной щелей монохроматора при записи спектров фосфоресценции была не более 1 мм.

Ошибка в определении максимума 0-0 полосы в спектре сенсибилизированной фосфоресценции не превышала 5 ![]() .
.
При изучении кинетики сенсибилизированной фосфоресценции для отделения её от фосфоресценции донора использовались электромеханические затворы, управляемые с помощью электронных реле времени. Время срабатывания затворов не превышало 5 мс. Задержка во времени между началом регистрации сенсибилизированной фосфоресценции и прекращением возбуждения донора изменялась от 0,1 до 30 с.
Регистрирующая часть установки включала в себя двухкоординатный графопостроитель Н-307 при записи спектров излучения и кинетики фосфоресценции молекул акцептора. При исследовании кинетики фосфоресценции молекул донора двухкоординатный графопостроитель заменялся на универсальный запоминающий осциллограф С8-13. Для согласования входного сопротивления самописца и выходного сопротивления фотоэлектронного умножителя использовался катодный повторитель, постоянную времени которого можно было изменять от 0,01 до 1,0 с. Линейность работы усилителя постоянного тока проверялась при подаче на ФЭУ светового потока регулируемого изменением входной щели монохроматора. Механическая постоянная времени графопостроителя не превышала 0,03 с.
Кинетические кривые, полученные с помощью графопостроителя или осциллографа перестраивались в полулогарифмическом масштабе, из которого и определялось время разгорания или время затухания фосфоресценции.
Величина погрешности при определении t в экспериментах обуславливалась флуктуациями фототока, нелинейностью усилителя, погрешностью блока временной развёртки и механической постоянной самописца. Три последних источника погрешностей по данным многократных проверок могли дать в сумме систематическую ошибку не более 1,0 %. Для уменьшения влияния флуктуаций фототока измерения повторялись 5-10 раз и общая ошибка в каждом конкретном случае находилась из среднего значения t с учётом возможной систематической ошибки. С учётом вышеизложенного, ошибка при измерении времени затухания сенсибилизированной фосфоресценции не превышала 0,05 с, а времени разгорания – 0,1 с. Большее значение ошибки при измерении времени разгорания обусловлено тем, что флуктуации светового потока источника света влияют на точность измерения времени разгорания и не влияют на точность измерения времени затухания. Ошибка при измерении времени затухания фосфоресценции донора не превышала 0,5 мс.
В температурных исследованиях для уменьшения продольного градиента температуры кварцевая кювета помещалась в медную толстостенную трубочку. При этом для исследования с помощью диафрагмы выделялся участок высотой 2 мм, где находился в образце один из концов дифференциальной термопары. Толщина кюветы была равна 0,3 мм, диаметр – 2 мм. С целью уменьшения влияния поперечного градиента температуры контрольные опыты проводились в лопаточкообразной кювете, толщина исследуемого слоя в которой около 0,5 мм.
Измерение температуры производилось с помощью медь-константановой термопары, проградуированной по точкам плавления н.-парафинов. Один спай термопары находился в сосуде Дьюара с жидким азотом, а второй помещался непосредственно в раствор перед замораживанием. В качестве измерительного прибора использовался гальванометр М-95, с ценой деления 0,01 мВ/дел. Ошибка при измерении температуры не превышала 3 К.
При исследовании температурной зависимости интенсивности и времени затухания сенсибилизированной фосфоресценции нагревание образца происходило в результате испарения азота под образцом. Скорость изменения температуры при этом была около 3 град./мин.
При исследовании температурной зависимости интенсивности сенсибилизированной фосфоресценции спектральная ширина щели бралась максимальной для того, чтобы смещение максимума 0-0 полосы при изменении не превышала её. Это и тот факт, что при увеличении температуры распределение интенсивности в спектре сенсибилизированной фосфоресценции не изменяется, позволяло судить по изменению регистрируемой интенсивности в максимуме 0-0 полосы об изменении интегральной интенсивности.
Отжиг образца производился следующим образом. Полученный в результате быстрого замораживания образец нагревался от 77 К до определённой температуры из области 150-180 К и выдерживался при фиксированной температуре необходимое время (от 0,5 до 40 мин.). Затем образец помещался в жидкий азот, в котором и производилось измерение его люминесцентных характеристик при 77 К.
Для определения влияния отжига на интенсивность сенсибилизированной фосфоресценции записывались спектры фосфоресценции раствора до и после отжига и сравнивались их интегральные интенсивности. Следует заметить, что в этом случае результаты совпадали с результатами, полученными при регистрации сенсибилизированной фосфоресценции в максимуме 0-0 полосы с точностью до 10 %.
При исследовании зависимости интенсивности сенсибилизированной фосфоресценции от времени отжига при фиксированной температуре образец отжигался в течение времени Dt , затем измерялись его люминесцентные характеристики при температуре 77 К. После чего образец снова нагревался до температуры отжига и отжигался в течение времени Dt , в результате чего время его отжига составляло 2Dt . Затем снова измерялись его люминесцентные характеристики. Таким образом, процесс повторялся до тех пор, пока не прекращался рост интенсивности в результате отжига образца.
III . Влияние отжига на параметры фосфоресценции дибромдифенилоксида и аценафтена в н.-октане.
В данной главе представлены результаты исследования влияния отжига на спектры, кинетику и интенсивность сенсибилизированной фосфоресценции молекул акцептора и обычной фосфоресценции молекул донора.
Одной из задач дипломной работы было исследование изменения люминесцентных характеристик донорно-акцепторной пары в результате выдерживания образца при постоянной температуре. Вычислить энергию активации, а также произвести сравнение изменений люминесцентных параметров и активаций процессов, происходящих при отжиге для других донорно-акцепторных пар.
3.1. Обработка полученных данных.
Ранее установлено, что отжиг раствора для многих донорно-акцепторных пар может приводить к увеличению интенсивности фосфоресценции как донора, так и акцептора и к уменьшению времени затухания.
Результаты исследования влияния отжига на фосфоресценцию донора представлены на рис.3.1. График 1 характеризует фосфоресценцию дибромдифенилоксида до отжига, график 2 – после отжига. Из рисунка видно, что интенсивность фосфоресценции после отжига уменьшилась примерно в 2 раза, чем до отжига.
Так как были получены данные, что интенсивность фосфоресценции акцептора уменьшилась после отжига (причем в большее число раз нежели донора), то представляло особый интерес исследование сенсибилизированной фосфоресценции акцептора.
Полученные данные по фосфоресценции донорно-акцепторной смеси, а так же по времени затухания акцептора сведены в табл.3.1.
Табл.3.1. Влияние отжига на параметры фосфоресценции компонент донорно-акцепторной смеси дибромдифенилоксида и аценафтена в н.-октане.
| донор | акцептор | ||
| I / I 0 | I / I 0 | τ/τ0 | |
| До отжига | 1 | 1 | 1 |
| После отжига | 0,55 | 0,32 | 0,79 |
Рис.3.1.

Обозначим интенсивность сенсибилизированной фосфоресценции после быстрого замораживания образца до 77 К через I (0) . После отжига образца в течение определённого времени t при температуре Т и последующем охлаждении до 77 К интенсивность сенсибилизированной фосфоресценции обозначим через I ( t ) . Тогда D I ( t ) = I ( t ) – I (0) – означает прирост интенсивности сенсибилизированной фосфоресценции в процессе отжига образца в течение этого времени.
Можно предположить, что при фиксированной температуре Т прирост интенсивности D I ( t ) в зависимости от времени отжига происходит по закону, определяемому экспонентой:
DI (t ) = DI (¥){1-exp(-t /t )}, (3.1)
с характерным временем t , которое зависит от температуры отжига. DI (¥) - прирост интенсивности при длительном отжиге образца - t » t .
Экспериментально эта зависимость была проверена в данной работе для пары дибромдифенилоксид-аценафтен в н.-октане. На рис. 3.2 представлен график зависимости [DI (t ) - DI (¥)]/DI (¥) от t в полулогарифмическом масштабе.

Рис. 3.2. Зависимость интенсивности сенсибилизированной фосфоресценции аценафтена, донор – дибромдифенилоксид, в н.-октане от времени отжига при температурах: 1 – 167 К, 2 – 180 К, 3 – 195 К; СД =1.25×10-3 М , СА =1.25×10-3 М; z = ln [( D I( ¥)- D I( t))/ D I( ¥)].
Как видно из рисунка, экспериментальные точки хорошо укладываются на экспоненту (сплошная линия) с различными углами наклона, определяемыми температурой отжига. Величина, обратная тангенсу угланаклона прямых, соответствует характерному времени t процесса при данной температуре отжига.Для всех исследованных систем повышение температуры отжига раствора приводит к уменьшению характерного времени процесса нарастания.
Табл.3.1. Характерное время t процесса нарастания числа одиночных молекул акцептора, участвующих в переносе энергии в процессе отжига.
| Тотж , К | q , 1 / мин | |
Дибромдифенилоксид-аценафтен в н.-октане |
167 | 0,48 |
| 180 | 0,71 | |
| 195 | 1,67 |
Выше было показано, что в твёрдом теле подобные физические и химические процессы обычно характеризуются Аррениусовской зависимостью константы скорости процесса от температуры:
q(Т) = q( ¥ ) ехр (-Еак / RT ) (3.2)
где q( ¥ ) - предэкспоненциальный множитель, Еак - энергия активации процесса.
Соответственно для t :
t (Т) = (1/ q( ¥ )) ехр (Еак / RT ). (3.3)
Представляло интерес экспериментально проверить эту зависимость.
Прологарифмируем уравнение Аррениуса (48):
ln t = Еак /RT- ln [q( ¥ )]. (3.4)
Написав это уравнение для различных температур Т1 и Т2 и вычтя второе уравнение из первого, получим:
ln ( t 1 / t 2 )= Еак / R (1/ T 1 - 1/Т2 ). (3.5)
Если это уравнение справедливо, то на графике в координатах ln ( t 1 / t ) от (1/ T 1 - 1/Т) экспериментальные точки должны располагаться на прямой с тангенсом угла наклона, равнымЕак / R .
На рис. 3.3 представлена данная зависимость для пары дибромдифенилоксид-аценафтен в н.-октане. Как видно из рисунка, экспериментальные точки хорошо укладываются на экспоненту (сплошная линия). Это говорит об экспоненциальной зависимости характеристического времени процесса t от температуры. Следовательно, и константа скорости q физического процесса, происходящего при отжиге экспоненциально растёт с повышением температуры.
Таким образом, на основании этих экспериментальных данных можно утверждать, что физический процесс, приводящий к увеличению числа участвующих в переносе энергии мономерных молекул акцептора при отжиге описывается Аррениусовской зависимостью константы скорости процесса от температуры.
Величина тангенса угла наклона прямых q позволяет определить энергию активации процесса: Еак = R tg q .
ln(τ1 /τ)

Рис.3.3 . Зависимость характеристического времени процесса от температуры, для донорно-акцепторных пары дибромдифенилоксид-аценафтен в н.-октане (СД =1.25×10-3 М, СА =1.25×10-3 М).
Энергия активации этого процесса для данной донорно-акцепторной пары представляет величину 9 кДж/моль.
3.2. Основные результаты и выводы.
Результаты экспериментального исследования влияния отжига на параметры фосфоресценции молекул дибромдифенилоксид-аценафтен в замороженных н.-парафиновых растворах можно сформулировать следующим образом:
1. Интегральная интенсивность спектра фосфоресценции дибромдифенилоксида в н.-октане в присутствии акцептора после отжига при Т=167К уменьшилась в 2 раза.При этом так же наблюдается смещение максимума 0-0 полосы в спектре фосфоресценции донора в коротковолновую область на 1-2нм.
2. Интегральная интенсивность спектра фосфоресценции акцептора при отжиге также уменьшается, причем в большее число раз, чем донора. Закон изменения интенсивности фосфоресценции акцептора от времени отжига носит экспоненциальный характер. Для всех исследованных систем повышение температуры отжига раствора приводит к уменьшению характерного времени процесса нарастания.
3. Закон зависимости константы скорости процесса носит аррениусовский характер:
q(Т) = q( ¥ ) ехр (-Еак / RT )
Энергия активации процесса для данной донорно-акцепторной пары представляет величину 9 кДж/моль.
4. Время затухания сенсибилизированной фосфоресценции акцептора после отжига уменьшается в 1,3 раза, при этом закон затухания становится не экспоненциальным. Вопрос о причинах данного процесса требует дальнейшего исследования.
Как показали результаты данной работы поведение пары дибромдифенилоксид-аценафтен при отжиге прямо противоположенные донорно-акцепторным парам бензофенон-аценафтен, бензофенон-нафталин, антрон-флуорен, интегральная интенсивность которых увеличивается в процессе отжига [13]. Такое поведение можно предположительно связать с тем, что растворимость дибромдифенилоксида в н.-октане намного меньше, чем остальных указанных веществ.
Если увеличение интенсивности фосфоресценции двухкомпонентных смесей авторы работ [13,25-27] объясняют распадом гетероассоциатов и снятием миграционно-ускоренного тушения, то в данном случае можно предположить, что из-за плохой растворимости происходит выкристаллизация примеси при температуре отжига, что приводит к уменьшению числа мономерных молекул.
Но не смотря на вышесказанное после замены донора энергии некоторые закономерности, полученные для бензофенона и других веществ, сохранились. Например, зависимость изменения интенсивности фосфоресценции акцептора от времени осталась экспоненциальной и зависимость константы скорости процесса от температуры носит аррениусовский характер.
Энергия активации процесса ответственного за рост интенсивности фосфоресценции смеси бензофенон-аценафтен Еак =40кДж/моль, а дибромдифенилоксид-аценафтен Еак =9кДж/моль.
Стоит отметить, что уменьшение времени затухания аценафтена после отжига запаздывает за спадом интенсивности сенсибилизированной фосфоресценции, т.е. τ/τo > I/Io . Если учесть наличие миграционно-ускоренного тушения на ассоциаты (микрокристаллы) в данном случае, то такое поведение данной зависимости стоило ожидать, так как время затухания τд донора во много раз больше времени затухания τак акцептора (порядка 1000 раз) и на акцептор данный процесс влияет сильнее.
Литература.
1. Климов В.В. Фотосинтез и биосфера // Соросовский образовательный журнал. – 1996. - № 8. – С. 6-13.
2. Миронов А.Ф. Фотодинамическая терапия рака – новый эффективный метод диагностики и лечения злокачественных опухолей // Соросовский образовательный журнал. – 1996. - № 8. – С. 32-39.
3. Зенькевич Э.И., Сагун Е.И., Кнюкшто В.Н. и др. Дезактивация S1 - и Т1 - состояний порфиринов и хлоринов при их взаимодействии с молекулярным кислородом в растворах // Ж. прикл. спектр. – 1996. – Т. 63. - № 4. – С. 599-612.
4. Копылова Т.Н. , Светличный В.А., Кузнецова Р.Т. и др. Флуоресцентные характеристики органических молекул при мощном импульсном лазерном возбуждении // Опт. и спектр. – 1998. – Т. 85. - № 5, - С. 778-782.
5. Бодунов Е.Н. Приближённые методы в теории безызлучательного переноса энергии локализованных возбуждений в неупорядоченных средах // Опт. и спектр. – 1993. – Т. 74. - № 3.- С. 518-551.
6. Королев В.В., Грицан Н.П., Хмельницкий И.В. и др. Определение параметров статического тушения фосфоресценции органических молекул по обменно-резонансному механизму // Хим. физ. – 1987. – Т. 6. - № 7. – С. 892-898.
7. Бурнштейн А.И. Концентрационное тушение некогерентных возбуждений в растворах // УФН. - 1984. - Т. 143. - № 4. - С. 533 - 600.
8. Бодунов Е.Н. Теоретические исследования спектральной миграции возбуждений в трехмерных средах. (Обзор) // Опт. и спектр. – 1998. – Т. 84. - № 3. – С. 405-430.
9. Журавлёв С.В., Левшин Н.В., Салецкий А.Н., Южаков В.И. О роли миграции между мономерными молекулами родаминовых красителей в концентрационном тушении люминесценции растворов // Опт. и спектр. – 1982. – Т. 53. - № 2. – С. 245-251.
10. Лёвшин Л.В., Салецкий А.М. Люминесценция и её измерения. Молекулярная люминесценция. - М.: Изд-во МГУ, 1989. - 272 с.
11. Ермолаев В.Л. Перенос энергии в органических системах с участием триплетного состояния. // УФН. - 1963. - Т. 80. - № 1. - С. 33-40.
12. Левшин Л.В., Салецкий А.М. Оптические методы исследования молекулярных систем. Ч.1. Молекулярная спектроскопия. – М.: Изд-во МГУ, 1994. - 320 с.
13. Дерябин и др. Особенности сенсибилизированной фосфоресценции аценафтена в кристаллах бензофенона / Дерябин М.И., Куликова О.И., Голубин М.А.; Ставроп. гос. пед. ун-т. - Ставрополь, 1996. - 10с. - Деп. в ВИНИТИ 03.04.96., № 1094 – В 96.
14. *Förster Th. // Ann. Phys. – 1948. - V. 2. - № 1-2. - Р. 55-75.
15. Dexter D.L. A Theory of Sensitized Luminescence in Solids // J. Chem. Phys. – 1953. – V. 21. - № 5. – P. 836-850.
16. Ермолаев В.Л. Сенсибилизированная фосфоресценция ароматических соединений (перенос энергии с триплетного уровня на триплетный) // Изв. АНСССР. – 1956. –Т. 20. - № 5. – С. 514-519.
17. Katayama Hideaki, Ifo Shinzaburo, Yamamoto Masahide Intramolecular triplet energy transfer of the system having donor and acceptor at the chain ends. II. The carbazole-naphthalene system // J. Phys. Chem. - 1992. – V. 96. - № 25. – Р. 10115-10119.
18. Haggquist Gregory W., Katayama Hideaki, Tsuchida Akira and oth. Intramolecular triplet energy transfer. III. A carbazole-naphthalene system having short chain length methylene spacer units // J. Phys. Chem. - 1993. – V. 97. - № 37. – Р. 9270-9273.
19. Engel Paul S., Horsey Douglas W., Scholz John N. аnd oth. Intramolecular triplet energy transfer in ester-linked bichromophorie aroalkanes and naphthalenes // J. Phys. Chem. - 1992. – V. 96. - № 19. – Р. 7524-7535.
20. Давыдов А.С. Электронные возбуждения и колебания решётки в молекулярных кристаллах// Изв. АН СССР. – 1970. –Т. 24. - № 3. – С. 483-489.
21. Петренко А.Н. Интегралы переноса триплетного возбуждения в линейных молекулярных кристаллах // Физ. твёрд. тела (С.-Перегбург). -1994. – Т. 36. - № 6. – С. 1784-1787.
22. Breenner H.C. Studies of triplet energy transter in molekular crystals by ODMR and high pressure techniques // Укр. физ. ж. – 1995. – Т. 40. - № 7. - С. 659-666.
23. Багнич С.А. Перколяция энергии электронного возбуждения по триплетным уровням бензальдегида в пористой золь-гелевой матрице // Опт. и спектр. – 1996. – Т. 80. - № 5. – С. 769-772.
24. Багнич С.А. Низкоэффективный транспорт триплетных возбуждений безальдегида в матрице пористое стекло – полиметилметакрилат // Опт. и спектр. – 1997. – Т. 82. - № 4. – С. 567-572.
25. Багнич С.А., Мельниченко И.М., Подденежный Е.Н. и др. Влияние матрицы на фосфоресценцию ароматических соединений в пористых золь-гелевых стеклах // Опт. и спектр. – 1995. – Т. 79. - № 6 – С. 936-941.
26. Багнич С.А., Богомолов В.Н., Курдюков Д.А. и др. Фосфоресценция ароматических соединений в пористой матрице натриево-боросиликатного стекла и взаимодействие со стенками пор // Физ. тв. тела (С-Петербург). – 1995. – Т. 37. - № 10. – С. 2979-2986.
27. Багнич С.А. Фосфоресценция бензофенона в условиях взаимодействия со стенками пористых матриц // Опт. и спектр. – 1996. – Т. 80. - №5. – С. 773-775.
28. Eremenko A.M., Smirnova N.P. Specific features of dye molecular luminescence in solid matrices // Funct. Mater. - 1996. – V. 3. - № 4. - P. 511-517.
29. Бегер В.Н., Сечкарев А.В . Влияние межмолекулярных взаимодействий в пространственно-неоднородных ансамблях молекул на безызлучательный перенос энергии электронного возбуждения // Ж. физ. химии. – 1995. – Т. 69. - № 3. – С. 567-572.
30. Бегер В.Н., Земский В.И. Особенности температурного тушения флуоресценции адсорбированных молекул органических красителей // Опт. и спектр. – 1993. – Т. 74. - № 3. – С. 552-556.
31. Сечкарев А.В., Земский В.И., Бегер В.Н. и др. Спектральные проявления фрактального распределения адсорбированных в порах молекул в условиях неоднородности межмолекулярных взаимодействий // Ж. физ. химии. – 1992. – Т. 66. - №2. – С. 329-334.
32. Бегер В.Н., Колесников Ю.Л., Сечкарев А.В. Особенности концентрационного тушения флуоресценции молекул красителей, адсорбируемых неоднородной поверхностью диоксида кремния // Опт. и спектр. – 1995. – Т. 78. - № 2. – С. 249-253.
33. Осипов В.В., Самойленко Ю.Я., Риттер А.Я. Существование динамического и статического механизмов тушения флуоресценции в адсорбируемом слое // Химия высоких энергий. – 1995. – Т. 29. - № 5. – С. 363-367.
34. Горяев М.А. Спектральная зависимость квантового выхода люминесценции адсорбированных красителей // Опт. и спектр. – 1997. – Т. 82. - №5. – С. 781-783.
35. Гобов Г.В., Конашенко В.И., Нурмухаметов Р.Н. Триплет-триплетный перенос энергии в условиях эффекта Шпольского // Опт. и спектр. – 1976. – Т. 40. - № 2. – С. 406-408.
36. Гобов Г.В., Конашенко В.И. Триплет-триплетный перенос энергии в условиях эффекта Шпольского // Ж. прикл. спектр. – 1978. – Т. 28. - № 4. – С. 663-667.
37. Гобов Г.В., Юденков В.В. Спектры сенсибилизированной фосфоресценции дифениленоксида в бинарных растворителях при 77 К // Электронно-колебательные спектры некоторых ароматических соединений. – Смоленск, 1975. – С. 20-23.
38. Гобов Г.В., Конашенко В.И. Спектры сенсибилизированной фосфоресценции кристаллических растворов при 77 К // Опт. и спектр. – 1977. – Т. 43. - № 6. – С. 1054-1059.
39. Гребенщиков Д.М., Дерябин М.И. Двухэкспоненциальное затухание сенсибилизированной фосфоресценции органических молекул в растворах при 77 К // Хим. физ. – 1989. – Т. 8. - № 12. – С. 1615-1618.
40. Вавилов С.И. Теория концентрационного тушения флуоресценции растворов // Собр. соч.– М.: изд. АН СССР, 1952. – Т. 2. - С. 122-130.
41. Бодунов Е.Н., Цвирко М.П. Расчёт оптимальной концентрации активаторов, обеспечивающих максимальный выход сенсибилизированной люминесценции в двухкомпонентных средах // Опт. и спектр. – 1992. – Т. 72. - № 4. – С. 884-888.
42. Бодунов Е.Н., Берберан-Сентуш М.Н., Мартиню Ж.М.Г. и др. Расчёт квантового выхода люминесценции при прыжковом механизме тушения методом Монте-Карло // Опт. и спектр. – 1996. – Т. 80. - № 6. – С. 909-912.
43. Бодунов Е.Н. Расчёт скорости концентрационного самотушения в рамках метода непрерывных во времени случайных блужданий // Опт. и спектр. – 1996. – Т. 81. - № 3. – С. 405-408.
44. Берберан-Сентуш М.Н., Бодунов Е.Н., Мартиню Ж.М.Г. Концентрационная зависимость квантового выхода сенсибилизированной люминесценции при переносе энергии с высоких возбужденных состояний // Опт. и спектр. – 1997. – Т. 83. - № 3. – С. 378-383.
45. Берберан-Сентуш М.Н., Бодунов Е.Н., Мартиню Ж.М.Г. Прыжковый механизм тушения люминесценции и диффузионное приближение // Опт. и спектр. – 1998. – Т. 85. - № 6. – С. 948-951.
46. Асенчук О.Д., Могильный В.В. Фотоиндуцированное структурирование и миграция энергии в ансамблях трехуровневых центрах при насыщении // Опт. и спектр. – 1995. – Т. 79. - № 5. – С. 800-804.
47. Багнич С.А., Дорохин А.В. Миграция энергии по триплетным уровням бензофенона в полиметилметанокрилате // Физ. тв. тела – 1991. – Т. 33. - № 5. – С. 1382-1386.
48. Сенаторова Н.Р., Левшин Л.В., Рыжиков Б.Д. Концентрационное тушение люминесценции в условиях неоднородного уширения электронных спектров молекул растворённого вещества // Ж. прикл. сектр. – 1979. – Т. 30. - № 4. – С. 658-661.
49. Рыжиков Б.Д., Левшин Л.В., Сенаторов Н.Р. О природе длинноволнового концентрационного смещения спектров люминесценции молекул примеси // Опт. и спектр. – 1978. – Т. 45. - № 2. – С. 282-287.
50. Гаевский А.С., Давыдова Н.А., Добровольская О.В. и др. Миграция энергии триплетных состояний пигментов типа хлорофилла и флуоресцеина // Изв. АН СССР – сер. физ. – 1980. – Т. 44. - № 4. – С. 783-788.
51. Бисенбаев А.К., Вязанкина Л.А., Мукушев Б.Т. и др. Исследования процессов ассоциации молекул красителей в водных растворах полиэлектролитов // Ж. прикл. спектр. – 1994. – Т. 60. - № 5-6 – С. 406-410.
52. Низамов Н., Хидирова Т.Ш., Захидов У. и др. Люминесценция ассоциированных молекул и комплексов органических красителей в растворах // Изв. АН СССР – сер. физ. – 1990. – Т. 54. - № 3. – С. 502-506.
53. Низамов Н., Хидирова Т.Ш., Юнусова М . Люминесценция разнородных димеров некоторых полиметиновых красителей в дихлорэтане // Ж. прикл. спектр. – 1991. – Т. 55. - № 5. – С. 881-884.
54. Низамов Н., Умаров К.У., Атаходжаев А.К . Спектроскопическое исследование межмолекулярных взаимодействий в растворах пиронина G и новометиленового голубого // Ж. прикл. спектр. – 1979. – Т. 30. - № 4. – С. 651-657.
55. Спектроскопия внутри- и межмолекулярных взаимодействий. / Под ред. Н. Г. Бахшиева. - вып. 2. – Л.: Изд. ЛГУ, 1978г. – 212 с.
56. *Левшин Л.В., Рева М.Г., Рыжиков Б.Д. // Вестник МГУ. - Сер. физика, астрономия. - 1981. - Т. 22. - № 4. - С. 75.
57. Журавлёв С.В., Левшин Л.В., Салецкий А.М. и др. Миграция электронного возбуждения в смешанных растворах красителей // Опт. и спектр. – 1984. – Т. 56. - № 6. – С. 1044- 1048.
58. Сверчков С.Е., Сверчков Ю.Е. Влияние структуры матрицы на скорость тушения люминесценции примесных центров в теории прыжковой миграции // Опт. и спектр. – 1992. – Т. 73. - № 3. – С. 488-492.
59. Соловьёв А.Н., Южаков В.И. Влияние комплексообразования на спектральные и люминесцентные характеристики растворов аминокумаринов // Изв. АН СССР. - Сер. физ.– 1990. –Т. 54. - № 3. – С. 513-517.
60. Шпольский Э.В . Проблемы происхождения и структуры квазилинейчатых спектров органических соединений при низких температурах // УФН. – 1962. – Т. 77. - № 2. – С. 321-336.
61. Davydov А .S . The radiationless transfer of energy of electronic excitation between impurity molecules in crystals // Phys. Stat. Solidi. – 1968. - V. 30. - № 1. - C. 357-366.
62. Brandon R., Gerkin R., Hutchison C. Electron magnetic resonance of triplet states and the detection of energy transfer in crystals // J. Chem. Phis., 1962, V. 37, № 2, Р. 447-448.
63. Сапунов В.В., Егорова Г.Д. Влияние температуры на некоторые бимолекулярные процессы с участием порфиринов и металлопорфиринов в водных растврах // Ж. прикл. спектр. – 1993. – Т. 59. - № 1-2. – С. 54-60.
64. Вавилов С.И. Собр. соч. - М.: Изд-во АН СССР, 1954. – Т. 1. – 450 с.
65. Химическая энциклопедия: В 5 т.: / Под ред. И. Л. Кнунянца. и др. - М.: Большая Российская энцикл., 1990. – Т. 2. - С. 631-635.
66. Борисевич Н.А., Казберук Д.В., Лысак Н.А. и др . Фотофизические и фотохимические релаксационные процессы в ароматических кетонах // Изв. АН СССР. - сер. физич. – 1990. - Т. 54. - № 3. - С. 370-376.
67. Головченко В.П., Файдыш А.Н., Кольчинский М.З . Влияние структуры решётки на фосфоресценцию чистых и примесных кристаллов бензофенона // Изв. АН СССР – сер. физич. – 1970. – Т. 34. - № 3. - С. 589-593.
68. Мамедов Х.И . Спектры поглощения, флуоресценции и фосфоресценции аценафтена в парафиновых растворителях // Изв. АНСССР – сер. физич. – 1965. – Т. 29. - № 8. - С. 1404-1406.
69. Dekkers J.J. Hoornweg G. Ph., Maclean C. and oth . Some characteristic features of Shpolskii spectra fluorescence spectra of acenaphthene in n-alkane matrices // J. of mol. spectr. - 1977. – V. 68. – P. 56-67.
70. Дерябин М.И., Дзарагазова Т.П., Падалка В.В. и др. Температурная зависимость спектров фосфоресценции аценафтена в матрицах н.-гексана // Вестник Ставропльского гос. пед. ун-та. – 1995. - № 2. - С. 116-119.
71. Борисевич Н.А., Водоватов Л.Б., Дьяченко Г.Г. и др. Колебательная структура уровней свободных молекул аценафтена в основном и возбуждённом электронных состояниях // Оп. и спектр. – 1996. - Т. 81. - № 5. – С. 757-761.
72. Доброхотова В.К., Кульчицкий В.А., Набойкин Ю.В. Спектры замороженных растворов двух примесей при 77К// Известия АН СССР. Серия физическая. – 1963. – Т.27. – №6. – С.690–692.
73. Климова Л.А., Нерсесова Г.Н. Спектры флуоресценции и поглощения бинарных смесей ароматических углеводородов в замороженных кристаллических растворах// Журнал прикладной спектроскопии. – 1965. – Т.2. – №1. – С.45–50.
74. Глядковский В.И., Климова Л.А., Нерсесова Г.Н. Спектроскопия смесей ароматических углеводородов в замороженных кристаллических растворах// Оптика и спектроскопия. – 1967. – Т.23. – №3. – С.407 – 413.
75. Cadas J.P., Courpron C., Lochet R. Transfersts á energie entre entre éħdts triplets miltien cristallin a 77K// CR.–1962.–V.254. – №4. – P.2490 – 2492.
76. Rouset A., Lochet R., Cadas J.P. Transferts á energie entre niveux triplets de la benzophenone et du naphtaline cristallisesa 77K// J. Phys.–1963.–V.24, №2. – P.2141–2143.
77. Гребенщиков Д.М., Блужин В.Б., Дзарагазова Т.П. и др. Т-Т перенос энергии между разными примесными центрами в матрицах Шпольского// Современные аспекты тонкоструктурной и селективной спектроскопии. – М.: 1984. – С. 27–31.
78. Расколодько В.Г., Файдыш А.Н. Спектры фосфоресценции и миграция энергии триплетного уровня в кристаллах бензофенона// Известия АН СССР. Серия физическая. – 1965. – Т.29. – №8. – С. 1309–1312.
79. Болотникова Т.Н., Наумова Т.М. К вопросу о концентрационной зависимости квазилинейчатых спектров фосфоресценции// Оптика и спектроскопия. – 1963. – Т.25. – №3. – С. 460 – 463.
80. Артюхов В.Я., Майер Г.В., Риб Н.Р.. Квантово-химическое исследование триплет-триплетного переноса энергии электронного возбуждения в бихромоформных молекулярных системах // Оптика и спектроскопия. – 1997. – Т.83. – №5. – С.743 – 748.
81. Spectroscopy and Excitation Dynamics of Condensed Molecular System / Eds. Agranovich V.H., Hochstraser R.M. – Amsterdam: North – Holland, 1983. Спектроскопияидинамикавозбужденийвконденсированныхмолекулярныхсистемах / Подред. Аграновича В.М. и Хохштрассера Р.М. – М, 1987 – 492с.
82. Мак-Глин С., Адзуми Т., Киносита М. Молекулярная спектроскопия триплетного состояния. – М.: Мир, 1972 – 448с.
83. Осадько И.С. Селективная спектроскопия одиночных молекул. – М.: Физматлит, 2000 – 319с.
84. Агранович В.М., Галанин М.Д. Перенос энергии электронного возбуждения в конденсированных средах. – М.: Наука, 1978 – 384с.
85. Ландау Л.Д., Лифшиц Е.М. Квантовая механика. – М.: Изд-во физико-математической литературы, 1963. – 704 с.
86. Inokuti M. Hirayama F. Influence of energy transfer by the exchange mecanism on donor luminescence // J. Chem. Phys. – 1965. – V.43. – №6. – P.1978 – 1989.
87. Медведов Э.С., Ошеров В.И. Теория безызлучательных переходов в многоатомных молекулах. – М.: Наука, 1977. – С.7–59.
88. Теренин А.Н. Фотоника молекул красителей. – Л.: Наука, 1967 – 616 с.
89. Паркер С. Фотолюминесценция растворов. – М.: Мир, – 1972 – 511с.
90. Красновский А.А. Фотохимия хлорофилла и его аналогов/ В сб. элементарные фотопроцессы в молекулах – М.: Наука. – 1966. – С. 213 – 242.
91. Портер Дж. Профессор Александр Теренин (1896 – 1967) – пионер фотохимии. К 100 – летию со дня рождения// Оптика и спектроскопия. – 1997. – Т.83. – №4. – С. 534 – 538.
92. Гурвич А.М. Введение в физическую химию кристаллофосфоров – М.: Высшая школа, 1982 – 376 с.