| Скачать .docx |
Курсовая работа: Методы химической идентификации
ФЕДЕРАЛЬНОЕ АГЕНТСТВО ПО ОБРАЗОВАНИЮ
ГОУ ВПО «Воронежский государственный технический университет»
КУРСОВАЯ РАБОТА
по дисциплине «Теоретические основы прогрессивных технологий»
Тема «Методы химической идентификации»
Выполнила студентка группы эк-091 Попова В.А.
Руководитель: Усачева Л.В.
Воронеж 2010
Содержание
1. Химическая идентификация………………………………………….. 3
1.1 Качественный анализ……………………………………………..10
1.2 Количественный анализ………………………………………….16
2. Методы количественного анализа……………………………………19
2.1 Амперметрическое титрование…………………………………..19
2.2 Потенциаметрическое титрование……………………………….21
2.3 Кислотно0основное титрование………………………………….24
2.4 Комплекснометрическое титрование…………………………….25
2.5 Титрование по методу осаждения………………………………..25
2.6 Окислительно-восстановительное титрование………….............26
3. Заключение…………………………………………………………….27
4. Список литературы……………………………………………………28
1. Химическая идентификация.
Химическая идентификация – это установление вида и состояния молекул, ионов, радикалов, атомов и других частиц на основе сопоставления экспериментальных данных с соответствующими справочными данными для известных частиц. Идентификация — установление тождества неизвестного соединения с другим известным.
Для этого сопоставляют физико-химические константы, свойства и реакции обоих веществ. Перед идентификацией вещества тщательно очищают, проводят предварительное его исследование: сопоставляют агрегатное состояние, цвет, вязкость, испытывают на растворимость в воде, органических растворителях, основаниях и кислотах, определяют горючесть и другие свойства .Например, в молекулярном анализе идентификация - установление химической формулы соединений или ее важнейших фрагментов. Идентификация - цель качественного анализа, который, как правило, предшествует количественным определениям.
Ошибки в идентификации компонентов пробы приводят к неправильным результатам количественного анализа. При идентификации определяют комплекс физико-химических свойств вещества: плотность, вязкость, температуры фазовых переходов, растворимость, показатели преломления, потенциалы ионизации, эмиссионные и абсорбционные спектры, индексы хроматографические удерживания, специфические химические реакции и другие. Иногда получают производные идентифицируемого вещества и сравнивают их свойства (например, температуры плавления) со свойствами соответствующих производных известного соединения.
Идентификация - важнейший и наиболее трудоемкий этап анализа многокомпонентных проб. Для ее облегчения создаются банки химико-аналитических данных. Универсальные аналитические приборы (атомно-абсорбц. спектрофотометры, хромато-масс-спектрометры, хроматографы с ИК Фурье детектированием, установки для рентгеноэлектронной спектроскопии и другие), обладающие большой разрешающей способностью и предназначенные для анализа многокомпонентных проб, снабжаются мини-ЭВМ, в которые вводится специализированная справочная химико-аналитическая информация. Логические операции, выполняемые химиком-аналитиком при идентификации, принципиально доступны для формализации, что позволяет создать системы искусств, интеллекта, использующие спектры ИК, ЯМР, ЭПР, хроматографические данные и т. п. Автоматизация идентификации возможна на основе принципов универсальной системы химического анализа, в которой каждое вещество имеет свой химико-аналитический код, представляющий собой совокупность детектируемых физико-химических свойств веществ.Качественный анализ характеризуется пределом обнаружения сухого вещества, т.е. минимальным количеством надежно идентифицируемого вещества, и предельной концентрацией раствора Сх,min. Эти вещества связаны друг с другом отношением
Сх,min-= предел обнаружения(мкг)/ объем раствора(мл) * 10
В качественном анализе применяются только такие реакции, пределы обнаружения которых не превышает 50 мкг. Существуют некоторые реакции, которые позволяют обнаружить то или иное вещество или ион в присутствии других веществ или других ионов. Такие реакции называются специфическими. Однако в большинстве случаев реакции обнаружения вещества не являются специфическими, поэтому мешающие идентификации вещества переводят в осадок, слабодиссоциирующее или комплексное соединение. Анализ неизвестного вещества проводят в определенной последовательности, при которой то или иное вещество идентифицирует после обнаружения и удаления мешающих анализу других веществ, т.е. применяют не только реакции обнаружения веществ, но и реакции отделения их друг от друга.
А) Свойства вещества зависят от его чистоты, рассмотрим понятие
чистоты веществ.Весьма практически важным вопросом, возникающим при различных химических работах, является вопрос о чистоте веществ. Определяя, например, состав какого-либо загрязненного примесями соединения путем его химического анализа, можно получить такие результаты, которые приведут к неверной формуле. Подобным же образом легко прийти к ошибочным выводам при изучении характера протекания химических реакций, т. е. получить ложное представление о свойствах участвующих в них элементов. Уже из приведенных примеров видно, что применяемые для химических работ вещества должны быть достаточно чистыми. К проверке чистоты вещества можно, вообще говоря, подойти с двух сторон: исходя из его состава или из его свойств. На практике часто параллельно используют оба подхода, так как результаты их хорошо дополняют друг друга. Принципиально простейшим (но не всегда легко выполнимым) методом проверки чистоты вещества, исходя из его состава, является количественный анализ: близкое совпадение найденного процентного содержания отдельных элементов с вычисленным по молекулярной формуле указывает обычно на отсутствие в изучаемом веществе значительных количеств примесей. Так как, однако, каждый анализ неизбежно связан с некоторыми неточностями, даже самые благоприятные его результаты не дают еще возможности говорить об отсутствии загрязнений. Характер последних удается большей частью заранее наметить исходя из природы контролируемого соединения и способа его получения. Отсутствие или наличие (а также количественное содержание) таких определенных примесей можно установить путем специальных проб. В этом и заключается другой часто применяемый метод контроля чистоты вещества исходя из его состава.
В основе контроля чистоты веществ по их свойствам лежит закон постоянства свойств (Пруст, 1806 г.): свойства чистого вещества не зависят от его происхождения и предыдущей обработки. Закон этот строго соблюдается только для газов и жидкостей, тогда как у твердых веществ может иметь место изменение некоторых свойств в зависимости от обработки. Поэтому применительно к твердым веществам законом постоянства свойств приходится пользоваться с известной осторожностью.
Из отдельных свойств веществ для контроля их чистоты лучше всего подходят те, которые могут быть изморены и выражены числом. Имея для какого-либо вещества точно установленный ряд характеризующих его констант (постоянных), на основании закона постоянства свойств следует ожидать, что точно такие же значения соответствующих констант будет иметь любой другой образец того же вещества, если он достаточно чист. Поэтому для контроля чистоты вещества нужно определить те или иные его константы и сравнить полученные результаты с уже имеющимися данными для заведомо чистого образца. Практически чаще всего определяют следующие константы: плотность, температуру плавления и температуру кипения.
Б) Молекулярный анализ-установление качества и количества состава химических соединений и их смесей.
При качественном анализе смеси химического соединения обычно предварительно разделяют различными методами (хроматографией, ректификацией, кристаллизацией, экстракцией, осаждением, термической диффузией и др.); затем для разделенных веществ определяют так называемые интегральные молекулярные признаки, к которым относятся молярная масса, суммарный элементный состав, плотность, растворимость, температуры фазовых переходов, показатели преломления, потенциалы ионизации, а также спектры поглощения электромагнитного излучения, масс-спектры и т. п. Эти характеристики химических соединений сопоставляют с соответствующими константами и спектрами образцов сравнения, устанавливают отсутствие депрессии (понижение и увеличение интервала) температуры плавления смеси идентифицируемого соединения и эталонного вещества (т.е. известного вещества, отождествляемого с исследуемым).
Часто определяют хроматографические характеристики веществ (индексы удерживания, объемы удерживания и др.); при этом одновременно идентифицируемое вещество отделяется от дркгих компонентов смеси. Идентификацию можно считать достоверной только в том случае, если совпадают несколько характеристик и констант идентифицируемого и эталонного веществ. Наиболее эффективны комбинированные методы: хромато-масс-спект-рометрия, сочетание высокоэффективной жидкостной хро-матографии и масс-спектрометрии, сочетание газо-жидкост-ной или жидкостной хроматографии и ИК спектроскопии с использованием преобразования Фурье и др. Соответствующие приборы снабжают микропроцессорами и соединяют с ЭВМ, содержащими банки спектральных и других аналитических данных. При качественном молекулярном анализе смеси веществ без предварительного разделения химического соединения обнаруживают по характерным химическим реакциям, спектрам поглощения, масс-спектрами и т.п.
В) Изотопный анализ — определение изотопного состава химического элемента. Изотопный анализ различных элементов можно реализовать на различных физических принципах. Наиболее распространенным является масс-спектрометрический метод, с помощью которого можно проводить изотопный анализ всех, без исключения, элементов периодической системы. Масс-спектрометры для определения изотопного состава должны быть очень точными. Для анализа изотопного состава легких элементов (углерод, водород, кислород, сера, азот и т. д.) используется ионизация электронным ударом. В этом случае годятся все методы ввода газовой фазы, как и в органических масс-спектрометрах.
Для анализа изотопов более тяжелых элементов используется термоионизация или ионизация в индуктивно-связанной плазме. Во многих типах изотопных масс-спектрометров используются магнитные масс-анализаторы. Важнейшими техническими характеристиками масс-спектрометров являются чувствительность, динамический диапазон, разрешение, скорость сканирования.
Оптические методы изотопного анализа (напр., абсорбционные на основе диодных лазеров) в ряде случаев позволяют избавиться от изобарных наложений (то есть помех при совпадении масс изотопов или изотопомеров различных элементов), которые часто препятствуют измерениям, проводимым при помощи масс-спектрометров.
Г) Фазовый анализ - определение химического состава и количества отдельных фаз в гетерогенных системах или индивидуальных форм соединения элементов в рудах, сплавах, полупроводниках и др. Объектом фазового анализа всегда является твердое тело.
Название "фазовый анализ" стало доминирующим, хотя некоторые авторы продолжают использовать другие термины: вещественный, рациональный, композиционный, локально-распределительный анализ. Обилие названий - следствие исторического процесса становления фазового анализа. Он возник из практических потребностей металлургии и металловедения, с одной стороны, и горно-обогатительного производства - с другой. Позднее фазовый анализ стал необходим в технологии полупроводников, при экологических исследованиях и в производстве пищевых продуктов.Основным методом фазового анализа в горной промышленности был процесс избирательного химического растворения с помощью K-T, щелочей, солей, окислительно-восстановительных реагентов и комплексообразующих веществ. В этой области фазовый анализ использовали для разработки рациональных технологий флотационного разделения и обогащения горнохимического сырья, его гидрометаллургической обработки. При этом, прежде всего стояли задачи идентификации, выявления и разделения различных оксидных или сульфидных соединений нескольких металлов или одного металла в разных степенях окисления. Причем эти соединения могли быть не только нативными (исходными) фазами - минералами, но и виртуальными (промежуточными, т. е. изолируемыми в ходе анализа, как, напр., индивидуальные оксиды, выделяемые из сложных природных соединений при обработке реагентами). Поэтому такой анализ можно было считать фазовым более или менее условно, а по существу он был рациональным (т. е. служил основой рациональной технологии) и вещественным, т.е. направленным на выделение и определение данного сложного или простого вещества, независимо от того, составляет ли оно с самого начала определенную фазу.
Предметом аналитической химии является химическая идентификация( качественный анализ) и измерения(количественный анализ).
1.1Качественный анализ
Качественный анализ имеет своей целью обнаружение определенных веществ или их компонентов в анализируемом объекте. Обнаружение проводится путем идентификации веществ, то есть установления тождественности (одинаковости) АС анализируемого объекта и известных АС определяемых веществ в условиях применяемого метода анализа. Для этого данным методом предварительно исследуют эталонные вещества, в которых наличие определяемых веществ заведомо известно. Например, установлено, что присутствие спектральной линии с длиной волны 350,11 нм в эмиссионном спектре сплава, при возбуждении спектра электрической дугой, свидетельствует о наличии в сплаве бария; посинение водного раствора при добавлении к нему крахмала является АС на присутствие в нем I2 и наоборот.
В настоящее время качественный анализ выполняют инструментальными методами: спектральными, хроматографическими, электрохимическими и др. Химические методы используют на отдельных стадиях инструментальных (вскрытие пробы, разделение и концентрирование и др.), но иногда с помощью химического анализа можно получить результаты более просто и быстро, например, установить наличие двойных и тройных связей в непредельных углеводородах при пропускании их через бромную воду или водный раствор KMnO4 . При этом растворы теряют окраску.
Детально разработанный качественный химический анализ позволяет определять элементный (атомный), ионный, молекулярный (вещественный), функциональный, структурный и фазовый составы неорганических и органических веществ.
При анализе неорганических веществ основное значение имеют элементный и ионный анализы, так как знание элементного и ионного состава достаточно для установления вещественного состава неорганических веществ. Свойства органических веществ определяются их элементным составом, но также и структурой, наличием разнообразных функциональных групп. Поэтому анализ органических веществ имеет свою специфику.
Качественный химический анализ базируется на системе химических реакций, характерных для данного вещества - разделения, отделения и обнаружения.
К химическим реакциям в качественном анализе предъявляют следующие требования.
1. Реакция должна протекать практически мгновенно.
2. Реакция должна быть необратимой.
3. Реакция должна сопровождаться внешним эффектом (АС):
а) изменением окраски раствора;
б) образованием или растворением осадка;
в) выделением газообразных веществ;
г) окрашиванием пламени и др.
4. Реакция должна быть чувствительной и по возможности специфичной.
Реакции, позволяющие получить внешний эффект с определяемым веществом, называют аналитическими , а добавляемое для этого вещество - реагентом . Аналитические реакции, проводимые между твердыми веществами, относят к реакциям «сухим путем », а в растворах - «мокрым путем ».
К реакциям «сухим путем» относятся реакции, выполняемые путем растирания твердого исследуемого вещества с твердым реагентом, а также путем получения окрашенных стекол (перлов) при сплавлении некоторых элементов с бурой.
Значительно чаще анализ проводят «мокрым путем», для чего анализируемое вещество переводят в раствор. Реакции с растворами могут выполняться пробирочным, капельным и микрокристалли-ческим методами. При пробирочном полумикроанализе его выполняют в пробирках вместимостью 2-5см3 . Для отделения осадков используют центрифугирование, а выпаривание ведут в фарфоровых чашечках или тиглях. Капельный анализ (Н.А. Тананаев, 1920 г.) осуществляют на фарфоровых пластинках или полосках фильтрованной бумаги, получая цветные реакции при добавлении к одной капле раствора вещества одной капли раствора реактива. Микрокристаллический анализ основан на обнаружении компонентов с помощью реакций, в результате которых образуются соединения с характерным цветом и формой кристаллов, наблюдаемых в микроскоп.
Для качественного химического анализа используют все известные типы реакций: кислотно-основные, окислительно-восстановительные, осаждения, комплексообразования и другие.
Качественный анализ растворов неорганических веществ сводится к обнаружению катионов и анионов. Для этого используют общие и частные реакции. Общие реакции дают сходный внешний эффект (АС) со многими ионами (например, образование катионами осадков сульфатов, карбонатов, фосфатов и т.д.), а частные - с 2-5 ионами. Чем меньше число ионов дают сходный АС, тем селективнее (избирательнее) считается реакция. Реакция называется специфической, когда позволяет обнаружить один ион в присутствии всех остальных.
Аммиак обнаруживают по запаху или по посинению красной лакмусовой бумажки, смоченной в воде и помещенной над пробиркой.
Селективность реакций можно повысить, изменяя их условия (рН) или применяя маскирование. Маскирование заключается в уменьшении концентрации мешающих ионов в растворе меньше предела их обнаружения, например путем их связывания в бесцветные комплексы.
Если состав анализируемого раствора несложен, то его после маскировки анализируют дробным способом. Он заключается в обнаружении в любой последовательности одного иона в присутствии всех остальных с помощью специфических реакций, которые проводят в отдельных порциях анализируемого раствора. Поскольку специфических реакций немного, то при анализе сложной ионной смеси используют систематический способ. Этот способ основан на разделении смеси на группы ионов со сходными химическими свойствами путем перевода их в осадки с помощью групповых реактивов, причем групповыми реактивами воздействуют на одну и ту же порцию анализируемого раствора по определенной системе, в строго определенной последовательности. Осадки отделяют друг от друга (например, центрифугированием), затем растворяют определенным образом и получают серию растворов, позволяющих в каждом обнаружить отдельный ион специфической реакцией на него.
Существует несколько систематических способов анализа, называемых по применяемым групповым реактивам: сероводородный, кислотно-основный, аммиачно-фосфатный и другие. Классический сероводородный способ основан на разделении катионов на 5 групп путем получения их сульфидов или сернистых соединений при воздействии H2 S, (NH4 )2 S, NaS в различных условиях.
Анионы при анализе в основном не мешают друг другу, поэтому групповые реактивы применяют не для разделения, а для проверки наличия или отсутствия той или иной группы анионов. Стройной классификации анионов на группы не существует.
Качественный химический анализ органических веществ подразделяют на элементный, функциональный, структурный и молекулярный.
Анализ начинают с предварительных испытаний органического вещества. Для твердых измеряют tплав. , для жидких - tкип , показатель преломления. Молярную массу определяют по понижению tзамерз или повышению tкип , то есть криоскопическим или эбулиоскопическим методами. Важной характеристикой является растворимость, на основе которой существуют классификационные схемы органических веществ. Например, если вещество не растворяется в Н2 О, но растворяется в 5%-ном растворе NaOH или NaHCO3 , то оно относится к группе веществ, в которую входят сильные органические кислоты, карбоновые кислоты с более чем шестью атомами углерода, фенолы с заместителями в орто - и параположениях, -дикетоны
Элементным анализом обнаруживают элементы, входящие в молекулы органических веществ (C, H, O, N, S, P, Cl, и др.). В большинстве случаев органическое вещество разлагают, продукты разложения растворяют и в полученном растворе определяют элементы как в неорганических веществах. Например, при обнаружении азота пробу сплавляют с металлическим калием, получая KCN, который обрабатывают FeSO4 , переводят в K4 [Fe(CN)6 ]. Добавляя к последнему раствор ионов Fe3+ , получают берлинскую лазурь Fe4 [Fe(CN)6 ]3 - (AC на присутствие N).
Функциональным анализом определяют тип функциональной группы. Например, реакцией с (NH4 )2 [Ce(NO3 )6 ] можно обнаружить спирт, а с помощью раствора KMnO4 отличить первичные, вторичные и третичные спирты. Первичные KMnO4 окисляет до альдегидов обесцвечиваясь, вторичные окисляет до кетонов, образуя MnO2 , а с третичными не реагирует.
Структурным анализом устанавливают структурную формулу органического вещества или ее отдельные структурные элементы (двойные и тройные связи, циклы и так далее).
Молекулярным анализом устанавливают целиком вещество. Например, фенол можно обнаружить реакцией с FeCl3 в пиридине. Чаще молекулярный анализ сводится к установлению полного состава соединения на основании данных об элементном, функциональном и структурном составе вещества. В настоящее время молекулярный анализ проводят в основном инструментальными методами.
При проведении расчета результатов анализа необходимо очень внимательно выполнять вычисления. Математическая погрешность, допущенная в числовых значениях, равносильна ошибке в анализе.
Числовые значения подразделяют на точные и приближенные. К точным, например, можно отнести число выполненных анализов, порядковый номер элемента в таблице Менделеева, к приближенным - измеренные значения массы или объема.
Значащими цифрами приближенного числа называют все его цифры, кроме нулей, стоящих слева от запятой, и нулей, стоящих справа после запятой. Нули, стоящие в середине числа, являются значащими. Например, в числе 427,205 - 6 значащих цифр; 0,00365 - 3 значащие цифры; 244,00 - 3 значащие цифры.
Точность вычислений определяется ГОСТ, ОСТ или ТУ на анализ. Если погрешность вычислений не оговорена заранее, то следует иметь в виду, что концентрация вычисляется до 4-ой значащей цифры после запятой, масса - до 4-го десятичного знака после запятой, массовая доля (процентное содержание) - до сотых долей.
Каждый результат анализа не может быть точнее, чем это позволяют измерительные приборы (поэтому в массе, выраженной в граммах, не может быть больше 4-5 знаков после запятой, т.е. больше точности аналитических весов 10-4 -10-5 г).
1.2Количественный анализ
Количественный анализ - определение содержания (массы, концентрации и т.п.) или количественных соотношений компонентов в анализируемом образце. Определяемыми компонентами могут быть атомы, молекулы, изотопы, функциональные группы, фазы и т. п. Обычно количественный анализ основан на использовании зависимости доступных измерению физических свойств изучаемого объекта или продукта его преобразования от состава.
Методика, или алгоритм проведения, количественный анализ подробно и в строгой последовательности регламентирует все стадии количественный анализ: отбор и подготовку пробы; переведение анализируемой части пробы в состояние, удобное для анализа; возбуждение и измерение аналитического сигнала - физической величины (оптической плотности, интенсивности спектральной линии, высоты полярографической волны, скорости счета импульсов в заданном канале гамма-спектрометра и т.д.), корреляционно связанной с содержанием определяемого компонента; построение градуировочной характеристики, описывающей эту связь; определение поправки контрольного опыта; расчет результата единичного определения; расчет результата анализа путем усреднения результатов единичных определений и др. Большинство методов количественного анализа относятся к сравнительным (относительным), в которых градуировочную характеристику строят с использованием образцов сравнения. В абсолютных методах количественный анализ (напр., гравиметрии, кулонометрии) образцами сравнения не пользуются. Большое значение имеют метрологические характеристики - закон распределения результатов параллельных определений, границы интервала определяемых содержаний, воспроизводимость, правильность, погрешности анализа
В основе количественного химического анализа лежит химическая реакция между определяемым веществом и веществом реагентом.
К химическим реакциям, применяемым в этом анализе, предъявляют следующие требования:
1) реакция должна протекать достаточно быстро и быть практически необратимой;
2) вещества, вступившие в реакцию, должны реагировать в строго определенных количественных соотношениях, т.е. реакция должна быть стехиометрической и не сопровождаться побочными реакциями;
3) в результате реакции должны получаться соединения с определенным молекулярным составом;
4) на ход реакции не должны оказывать влияние примеси, присутствующие в анализируемом веществе;
5) реакция должна позволять достаточно просто устанавливать момент ее окончания, а также массу продукта реакции или объем раствора реагента, затраченный на ее проведение.
Несмотря на требование необратимости, большинство аналитических реакций до конца не идут, поскольку продукты реакции взаимодействуют друг с другом с образованием исходных веществ. В начале химического обратимого процесса скорость прямой реакции максимальна, а обратной реакции равна нулю, но по мере прохождения процесса скорость прямой реакции уменьшается с уменьшением концентраций исходных веществ, а скорость обратной растет.
Зависимость скорости реакции от концентрации реагирующих веществ выражается законом действующих масс (К. Гульдберг, П. Вааге, 1867 г.): скорость химической реакции при данной температуре пропорциональна произведению концентраций реагирующих веществ, в степенях, равных стехио Константа скорости химической реакции - это ее скорость при единичных концентрациях реагирующих веществ. При постоянной температуре константа скорости зависит только от природы реагирующих веществ и не зависит от их концентрации, что позволяет сравнивать скорости различных реакций путем сравнения их констант. Зависимость К = f (Т) выражает уравнение Аррениуса ℓnK=A/T+B (A и В - константы), а также империческое правило Вант-Гоффа: при увеличении температуры на каждые 10°С скорость химической реакции увеличивается в 2…4 раза.
Состояние системы реагирующих веществ, при котором скорость прямой и обратной реакции равны между собой, называется химическим равновесием:
![]() ,
,
где квадратными скобками показаны концентрации реагирующих веществ в момент равновесия.
Константу КР называют константой химического равновесия, а уравнение для ее вычисления выражает ЗДМ для химического равновесия: при установившемся химическом равновесии отношение произведения концентрации продуктов к произведению концентрации реагирующих веществ, в степенях, соответствующим стехиометрическим коэффициентам, есть величина постоянная для данной реакции при определенных условиях.
Физический смысл КР в том, что она показывает во сколько раз V1 > V2 или в сторону какой реакции смещено равновесие. Для аналитических целей чаще всего используют реакции, имеющие большую величину КР и практически нацело смещенные в прямом направлении.
метрическим коэффициентам в уравнении реакции
Концентрация- это величина, показывающая количественное содержание одного вещества в другом в относительных единицах, таких, как:
- процент (%), выражающий число частей данного вещества на 100 частей другого (или всего) вещества;
- промилле (‰, рm) - на тысячу частей;
- пропромилле (‰0 , ррm) - на миллион частей;
- пробилле (рв) - на миллиард частей;
- кг/м³, г/см³, моль/дм³, кг/т и др.
2. Методы количественного анализа
2.1Амперометрическое титрование
Амперометрическое титрование, метод количественного анализа, основанный на волътамперометрии с линейной разверткой потенциала. Конечную точку титрования устанавливают по зависимости диффузионного тока Idпри постоянном потенциале Ес индикаторного электрода от объема V прибавленного титранта. Электрохимически активным веществом, обусловливающим измеряемый диффузионный ток, может быть определяемый компонент, титрант, продукт их взаимодействия или вещество ("индикатор"), дополнительно введенное в электролитическую ячейку. Выбор значения Ес производят по вольтамперограммам определяемого вещества и титранта. Титрантом служит раствор реагента - осадителя, окислителя, восстановителя или комплексообразующего вещества, концентрация которого на несколько порядков превышает концентрацию определяемого вещества, с которым он взаимодействует. Титрант прибавляют из микробюретки небольшими порциями, благодаря чему разбавлением исследуемого раствора можно пренебречь. Форма кривых титрования Id=f(V)зависит от того, какое вещество электроактивно до и после точки эквивалентности, но всегда эти кривые имеют по крайней мере две ветви, которые пересекаются в конечной точке (к. т.) титрования.
Вольтамперограммы (а) определяемого электроактивного в-ва и кривая его амперометрич. титрования (6).
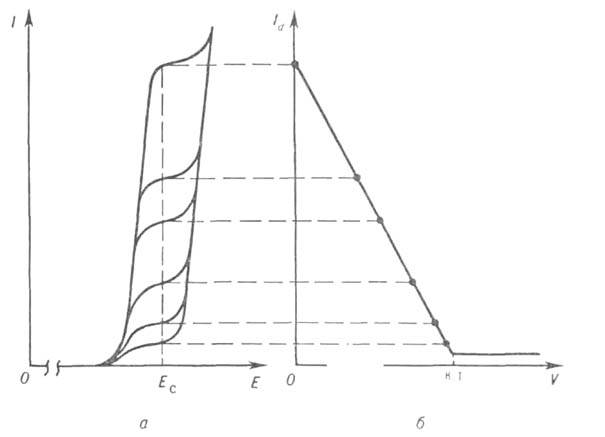
Обычно эти ветви прямолинейны. На рисунке в качестве примера приведена кривая титрования электроактивного вещества нелектроактивным реагентом. При последовательном титровании нескольких веществ на кривой титрования имеется соответствующее число точек перегиба. Важное преимущество амперометрического титрования перед вольтамперометрией - возможность определять не только электроактивные вещества, но и любые другие с применением электроактивных реагентов. В качестве поляризующегося индикаторного электрода применяют ртутный электрод, но чаще твердые электроды из благородного металла (обычно платины) или углеродного материала (графита, стеклоуглерода и др.). В амперометрическом титровании можно использовать два индикаторных электрода (без электрода сравнения), выполненных из одного и того же материала в виде пластин с одинаковыми относительно большими поветями (1 х 1 см). Этот вариант иногда неправильно называют биамперометрическим титрованием. Электроды погружают в титруемый раствор, содержащий два электроактивных вещества и индифферентный электролит. К электродам прикладывают напряжение, обеспечивающее электродные процессы одновременно на аноде и катоде двух разных веществ или сопряженных (окисленной и восстановленной) форм одного и того же вещества. Величину выбирают так, чтобы ток соответствовал предельному для обоих процессов. Роль индикаторного играет тот электрод, на котором электродный процесс обусловлен веществом, присутствующим в меньшей концентрации; второй электрод практически служит электродом сравнения. Иногда выбирают так, что оно мало и не соответствует предельному току электроактивных веществ; при этом ветви кривых титрования не прямолинейны, однако в к.т. ток цепи уменьшается до остаточного, или фонового, обусловленного, например, примесями (этот вариант ранее называли "амперометрическим титрованием до мертвой точки"). Нижние границы определяемых содержаний в амперометрическом титровании с одним индикаторным электродом 10-5 М, с двумя - 10-6 М. Амперометрическое титрование широко применяют для анализа органических и неорганических веществ, определения растворимости осадков, устойчивости комплексных соединений и т.д.
Электрохимические методы анализа основаны на измерении электрической проводимости, потенциалов, тока и других величин. Характерной особенностью при этом является электрический характер аналитического сигнала. Группа электрохимических методов анализа включает методы потенциометрии, кондуктометрии, амперометрии и др.
2.2Потенциометрическое титрование
Потенциометрическое титрование основано на определении точки эквивалентности по результатам потенциометрических измерений. Вблизи точки эквивалентности происходит резкое изменение (скачок) потенциала индикаторного электрода. Это наблюдается, конечно, лишь тогда когда хотя бы один из участников реакции титрования является участником электродного процесса. Так, например, титрование по методу кислотно-основного взаимодействия может быть выполнено со стеклянным электродом. Определение хлорида - с хлорсеребряным и т.д. Так же, как и в других титриметрических методах, реакции потенциометрического титрования должны протекать строго стехиометрически, иметь высокую скорость и идти до конца. Для потенциометрического титрования собирают цепь из индикаторного электрода в анализируемом растворе и электрода сравнения. В качестве электродов сравнения чаще всего применяют каломельный или хлорсеребряный.
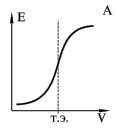
Определение точки эквивалентности
На рис. А представлена кривая титрования хлороводородной кислоты (HCl) гидроксидом натрия (NaOH). Она почти точно воспроизводит теоретическую кривую титрования сильной кислоты сильным основанием. Как видно, в точке эквивалентности происходит резкий скачок ЭДС, вызванный резким изменением потенциала индикаторного электрода. По этому скачку можно определить точку эквивалентности и потом рассчитать содержание хлороводородной кислоты.
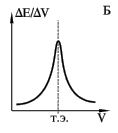
Для нахождения точки эквивалентности часто строят дифференциальную кривую в координатах dE/dV - V (рис. Б). На точку эквивалентности указывает максимум полученной кривой, а отсчет по оси абсцисс, соответствующий этому максимуму, дает объем титранта, израсходованного на титрование до точки эквивалентности. Определение точки эквивалентности по дифференциальной кривой значительно точнее, чем по простой зависимости E - V.
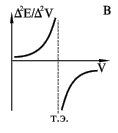
Поскольку производная функции, имеющей максимум, в точке максимума равна нулю, вторая производная потенциала по объему (d2E/dV2) в точке эквивалентности будет равна нулю. Это свойство также используется для нахождения точки эквивалентности (рис. В).
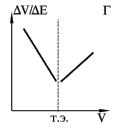
В простом и удобном методе Грана точка эквивалентности определяется по графику в координатах dV/dE-V. Перед точкой эквивалентности и после нее кривая Грана линейна, а сама точка эквивалентности находится как точка пересечения этих прямых (рис. Г). Достоинства и удобства метода Грана особенно заметны при анализе разбавленных растворов, позволяя определить точку эквивалентности с достаточной точностью вследствие линейности графика.
Виды потенциометрического титрования
2.3Кислотно-основное титрование
В кислотно-основном титровании в качестве индикаторного обычно используют стеклянный электрод, как правило, входящий в комплект серийно выпускаемых промышленностью pH-метров. Потенциометрический метод позволяет провести количественное определение компонентов в смеси кислот, если константы диссоциации различаются не менее чем на три порядка. Например, при титровании смеси, содержащей хлороводородную (HCl) и уксусную кислоты, на кривой титрования обнаруживается два скачка. Первый свидетельствует об окончании титрования HCl, второй скачок наблюдается при оттитровывании уксусной кислоты. Также несколько скачков имеют кривые титрования многоосновных кислот, константы диссоциации которых существенно различаются (хромовая, фосфорная и др.).
Широкие возможности анализа многокомпонентных смесей без разделения открывает применение неводных растворителей. Например, определение содержания хлороводородной и монохлоруксусной кислот в смеси титрованием водного раствора является сложной задачей в связи с трудностью обнаружения двух скачков титрования. При титровании в ацетоне оба скачка выражены достаточно четко и содержание каждой кислоты в смеси может быть рассчитано.
2.4Комплексонометрическое титрование
Потенциометрическое титрование катионов комплексоном III (ЭДТА) можно проводить с использованием в качестве индикаторного электрода соответствующего металла: титрование солей меди с медным электродом, солей цинка с цинковым и т.д. или подходящего ионоселективного электрода. Однако, многие металлические индикаторные электроды необратимы, а число ионоселективных электродов невелико.
Для комплексонометрических титрований может быть использован универсальный электрод Hg|HgY2- или Au(Hg)|HgY2- где Au(Hg) - амальгамированное золото; HgY2- - комплекс ртути с анионом этилендиаминтетрауксусной кислоты. С помощью ртутного электрода этого типа могут быть оттитрованы любые ионы, которые образуют с Y4- комплексы с константой устойчивости, не превышающей константу устойчивости ртутного комплекса. Это, например, ионы магния (Mg2+), кальция (Ca2+), кобальта (Co2+), никеля (Ni2+), меди (Cu2+), цинка (Zn2+) и др.
2.5Титрование по методу осаждения
Индикаторными электродами в методах потенциометрического титрования, использующих реакции осаждения, служат металлические или мембранные электроды, чувствительные к определяемому иону или иону-осадителю. Практически по методу осаждения могут быть определены катионы серебра, ртути, цинка, свинца, анионы хлора, брома, иода и некоторые другие. Смесь галогенидов, например I- и Cl-, может быть оттитрована без разделения нитратом серебра. Серебряный электрод позволяет фиксировать два скачка в ходе такого титрования. Первый скачок свидетельствует об оттитровывании иодид-иона и может быть использован для расчета содержания этого иона, второй скачок относится к окончанию осаждения хлорид-иона. По второму скачку можно рассчитать суммарное содержание галогенидов или концентрацию хлорид-иона, если концентрация иодид-иона будет известна из данных по титрованию до первого скачка.
2.6Окислительно-восстановительное титрование
Кривые окислительно-восстановительного титрования могут быть построены в координатах или pM - V (титранта) или E - V (титранта), если pM=-lg[M] ([M] - концентрация участника реакции, E - потенциал системы, V (титранта) - объем титранта. Кривые титрования первого типа представляют практический интерес, когда имеется индикаторный электрод, чувствительный к M. Кривые второго типа имеют более общее значение, так как любое окислительно-восстановительное титрование может быть проведено по измерению E с использованием индикаторного электрода из благородного металла, чаще всего платины.
Заключение
В настоящее время имеется широкий выбор различных аналитических методов и приемов. Независимо от того, как они подразделяются — на химические и инструментальные, качественные и количественные, весовые и объемные, оптические и электроаналитические, приложимые к органическим или неорганическим объектам, - все аналитические методы основаны на физических и (или) химических свойствах исследуемой системы. С разработкой новых приборов и методик анализа химик получает в распоряжение средства, позволяющие заняться еще более сложными проблемами. И наоборот, попытки решить сложные проблемы порождают потребность в разработке новых аналитических методов.
Анализ - это главное действующее лицо в любой научной проблеме, и, вероятно, неадекватная и неточная химическая идентификация привела к большему числу ошибочных научных результатов, чем любая другая причина. Отсюда вы можете заключить, насколько важно овладеть методами аналитической химии и уметь критически оценивать результаты анализов
Список литературы
1. Андрушко, Г.С. Химия. Химическая идентификация: Учеб. пособие / Г.С. Андрушко, О.В. Носова; М-во образования Рос. Федерации. Норил. индустр. ин-т. - Норильск : НИИ, 1999. - 65 с.
2. Васильев, В. П. Аналитическая химия : учеб. для студентов вузов, обучающихся по химико-технол. специальностям / В. П. Васильев. - 3-е изд., стер. - Москва : Дрофа, 2003.
3. Золотов, Ю. А. (гл. ред.). Физико-химические методы анализа / что это такое? // Журнал аналитической химии. - 2007. - Т. 62, N 10. - С. 1013.
4. Кристиан, Г. Аналитическаяхимия : в 2 томах / Г. Кристиан ; пер. с англ. А. В. Гармаша [и др.]. - Москва : Бином. Лаб. знаний, 2009.
5. Мухина, Е.А. Физико-химические методы анализа / Мухина, Е.А. . - М. : Химия, 1995. - 415с.
6. Пиментел, Д.К. Возможностихимиисегодняизавтра / Дж. Пиментел, Дж. Кунрод; Пер. с англ. В. А. Сипачева, Ю. А. Устынюка; Под ред. Ю. Д. Третьякова. - М.: Мир, 1992. - 288 с.
7. Притчина Е. А., Венедиктов А. Б. Химические методы идентификации и определения. Новосибирск: Новосибирский гос. ун-т, 2001.
8. Физико-химические методы анализа и контроля производства : Межвуз. науч.-темат. сборник / Дагестан. гос. ун-т им. В.И. Ленина ; Редкол.: О.А. Татаев (отв. ред.)и др. - Махачкала : Издательство Дагестан. ун-та, 1991.
9. Химическая идентификация веществ: Учеб. пособие / М-во образования РФ. С.-Петерб. гос. электротехн. ун-т "ЛЭТИ" ; [В.Ф. Иванов и др.]. - СПб. : ЛЭТИ, 2002. - 87 с.