| Скачать .docx |
Реферат: Определение компонентов ванн крашения кислотными красителями
Московский Государственный Текстильный Университет
им А.Н. Косыгина.
Кафедра аналитической, физической и коллоидной химии.
Отчет по учебно-исследовательской работе на тему: «Определение компонентов ванн крашения кислотными красителями».
Выполнили: студенты гр.26-99
Белая В.В,
Галимова Л.Р.
Проверил: доцент Чеснокова О.Я.
Москва 2002г.
СОДЕРЖАНИЕ.
| Задание на УИР |
3 стр |
| Введение |
4 стр |
| 1. Обзор литературы на тему «Методы определения красителей» |
7 стр |
| 2. Описание методов, установок и обработки результатов определений |
10 стр |
| 2.1.Установка для прямой фотометрии |
10 стр |
| 2.2. Установка для полуавтоматического потенциометрического титрования. |
12 стр |
| 2.3. Установка для высокочастотного титрования |
13 стр |
| 3. Результаты и их обсуждение |
16 стр |
| 3.1. Исследование условий фотометрического определения красителей в кислотной ванне крашения кислотными красителями |
16 стр |
| 3.2. Определение концентрации красителя в технологической ванне крашения кислотными красителями методом прямой фотометрии |
21 стр |
| 3.3. Исследование условий определения концентрации уксусной кислоты в кислотных ваннах крашения методом полуавтоматического потенциометрического титрования |
22 стр |
| 3.4. Определение концентрации уксусной кислоты в технологической ванне крашения кислотными красителями методом полуавтоматического потенциометрического титрования |
25 стр |
| 3.5. Исследование условий определения концентрации сульфата натрия в кислотных ваннах крашения методом высокочастотного титрования |
26 стр |
| 3.6. Определение концентрации сульфата натрия в технологической ванне крашения кислотными красителями методом высокочастотного титрования |
29 стр |
| Сводная таблица |
30 стр |
| Выводы |
31 стр |
| Список литературы |
33 стр |
Задание на УИР.
Тема: «Определение компонентов ванны крашения кислотными красителями».
Содержание задания
1. Составить обзор литературы на тему: «Методы определения красителей».
2. Исследовать условия определения и определить содержание: красителя методом прямой фотометрии, уксусной кислоты методом полуавтоматического потенциометрического титрования, Nа2 SO4 методом высокочастотного титрования.
3. Установить метрологические характеристики результатов определения.
Оценка УИР
ВВЕДЕНИЕ.
Красителями называются органические соединения, обладающие способностью интенсивно поглощать и преобразовывать энергию электромагнитных излучений (световую энергию) в видимой, ближних ультрафиолетовой и инфракрасных областях спектра и применяемые для придания (сообщения) этой способности другим телам[4].
Кислотными красителями называют водорастворимые соли органических кислот, главным образом сульфо- , реже – карбоновых кислот, иногда соли фенолов. В водных растворах диссоциируют с образованием цветных анионов красителя. Компенсиррующим катионом чаще всего является катион натрия Na+ , реже аммония NH4 + . В качестве хромофора используют моноазо−, диазо−, антрахиноновые и трифенилметановые структуры. В качестве ауксохромов в молекулу вводят –ОН; −NH2 ; −NHR и другие полярные электроно-донорные или электроно-акцепторные группы. Таким образом, чаще всего кислотные красители являются натриевыми солями сложных органических сульфокислот и имеют молекулярную массу 300−500[3]. Общая формула кислотных красителей выглядит следующим образом Кр SO3 - Na+ . К типичным представителям кислотных красителей относятся кислотный красный 2С:
и кислотный антрахиноновый ярко-зеленый Н2С:
По количеству марок в мировом ассортименте кислотные красители занимают 1 место. В отечественном ассортимете насчитывается более 60 наименований кислотных красителей, ими окрашивают белковые (шерсть и натуральный шелк) и полиамидные (ПА) волокна[1].
Кислотные красители отличаются яркостью и сочностью окрасок, присущих хромофорам достаточно простого строения, а в целом красители этого класса характеризуются широким спектром цвета. Они обладают сродством к волокнам, имеющим амфотерный характер (т.е. к шерстяным, шелковым и синтетическим полиамидным волокнам), и окрашивают их из водного раствора в присутствии кислот, вступая в солеобразование с молекулами этих веществ за счет содержания в них основных групп (-NH2 ), приобретающих положительный заряд (-NH3 + ). Отрицательно заряженный анион красителя КрSO3 - взаимодействует с волокном за счет ионных (солевых) связей. К целлюлозным волокнам кислотные красители сродства не имеют.
Недостатками кислотных красителей являются:
· способность ионной связи к гидролизу в водных растворах и, следовательно, невысокая устойчивость окраски к мокрым обработкам;
· мгновенное проявление сродства красителя к волокну приводит к его неровному окрашиванию.
Крашение кислотными красителями проводят в присутствии нейтрального электролита Na2 SO4 , который способствует более равномерной окраске (анионы SO4 -2 конкурируют с анионами красителя за положительные активные центры волокна).
По ровняющей способности кислотные красители делят на 3 группы: хорошо, средне и плохо ровняющие. Для улучшения ровноты крашения кислотными красителями белковых, полиамидных и других волокон, содержащих в своем составе основные группы −NH2, >HN, варьируют рН, температуру, вводят в красильную ванну электролиты, ТВВ и ПАВ, снижающие скорость выбирания красителей.
Кислотные красители различаются между собой по скорости перехода на полиамидное волокно, по выравнивающей способности и прочности получаемых окрасок к мокрым обработкам. Красители делят на хорошо, средне и плохо выравнивающие красители.
Хорошо выравнивающиеся красители обладают малым сродством к волокну, высокой диффузионной подвижностью, они окрашивают с высокой равномерностью, но малоустойчивы к мокрым обработкам.
Средне выравнивающиеся красители имеют хорошую устойчивость окраски к мокрым обработкам.
Плохо выравнивающиеся красители характеризуются низкой диффузионной подвижностью и высоким сродством к волокну. Они обладают высокой устойчивостью к мокрой обработке, но получаемая окраска недостаточно равномерная. Эта группа красителей является наиболее ценной, так как равномерность окраски можно повысить путем применения выравнивателей.
В зависимости от ровняющей способности красителей изменяется рецептура и режим крашения. Красильная ванна содержит % массы волокна: красителя 3−6, глауберовой соли 10, серной кислоты 2−4 (ρ=1,84; рН=2-4) для хорошо выравнивающих. Для средневыравнивающих вместо минеральной кислоты вводят 30%−ную уксусную кислоту 3−5% (рН=4-6), а в случае использования плоховыравнивающих красителей − сульфат или ацетат аммония 3−5% (рН=6-7).
Прочность окраски кислотными красителями шелка ниже, чем на шерсти, и ниже, чем на шелке, окрашенном прямыми и, конечно, активными красителями. Полиамидные волокна имеют относительно низкую кислотную емкость, но достаточно интенсивно окрашиваются кислотными красителями по режимам, близким к крашению шерсти, окраски отличаются высокой устойчивостью к мокрым обработкам.
Сильные минеральные кислоты в крашении полиамидных волокон не используются во избежании кислотного гидролиза. В случае хорошовыравнивающих красителей серную кислоту заменяют на муравьиную (85% −ная, 2 −4% массы волокна).
Определение класса красителя, используемого для крашения? имеет большое значение для достижения желаемого оттенка, способа заключительной обработки волокна.
Поскольку состав красильной ванны влияет на качество крашения, то целью нашей работы было исследование условий определения и установление содержания красителя методом прямой фотометрии, уксусной кислоты методом полуавтоматического потенциометрического титрования, сульфата натрия методом высокочастотного титрования в кислотных ваннах крашения.
Состав технологического раствора:
| Кислотный краситель |
0,1 - 0,4 г/л |
| Nа2 SO4 (М=142,04) |
2 - 10 г/л |
| CH3 COOH (М=60,05 г/моль) |
0.2 - 0,4 г/л |
| ПАВ(смачиватель Ва(CH3 COO)2 ) |
0.2 - 0,5 г/л |
1.ОБЗОР ЛИТЕРАТУРЫ НА ТЕМУ: «МЕТОДЫ ОПРЕДЕЛЕНИЯ КРАСИТЕЛЕЙ»
Количесвенные методы анализа красителей имеют как научное, так и практическое значение. Количественный анализ самих красителей, их растворов или дисперсий, а также окрашенных волокон полезен для оценки новых красителей, разработки процессов крашения, исследований прочности красителей или механизмов крашения. При крашении количественный анализ также важен для контроля качества продукции.
Для успешной разработки эффективных процессов крашения требуется количественная оценка степени использования красителя и точное понимание соответствующих механизмов. При крашении краситель либо адсорбируется на волокне или в нем, либо остается неиспользованным; при крашении до истощения ванны часть красителя все же остается в красильном растворе. Изучение процессов крашения становиться более значимым, когда измеряют каждую из этих частей.
Определение содержания красителей в красильных и плюсовых ваннах.
Содержание красителей в ваннах определяют колориметрическим методом сравнения с помощью фотоэлектроколориметра оптической плотности исследуемого раствора и раствора с известной концентрацией красителя.
Определение водорастворимых красителей в водных красильных ваннах не представляет затруднений, если контролируются факторы, влияющие на поглощение красителя (значение рh, ионная сила). Если же красильная ванна содержит нерастворимые в воде красители или вспомогательные вещества, то для получения однородной пробы необходимо добавлять к ванне смешивающейся с водой растворитель, такой, как ацетон, метанол, пиридин или ДМФ[3].
Определение красителей на поверхности волокна
Количество красителя, осажденного на поверхности волокна, может быть определено экстракцией окрашенного волокна таким растворителем, который растворяет краситель, но не вызывает набухания волокна, так что сорбированный внутри волокна краситель не экстрагируется. Определение количества красителя на поверхности волокна проводят при крашении до истощения ванны, для оценки миграции красителя с волокна в слой аппретуры на поверхности волокна, для оценки фиксации красителя в непрерывных процессах крашения и т.д.
Фиксацию красителя лучше всего определять, экстрагируя нефиксированный (поверхностный) и фиксированный (сорбированный) краситель в одной и той же пробе.
Определение красителя, сорбированного волокном, по спектрам отражения
Количественный анализ красителей, сорбированных волокном, по спектрам отражения − быстрый метод, не разрушающий образца, в принципе сходен с визуальной оценкой. Методы измерения отраженного света оказались полезными при контроле качества промышленной продукции, подборе цвета, в контрольных системах автоматизированных процессов.
Точность определения красителя в растворе с помощью проходящего света в два раза выше таковой для определения красителя на волокне по отраженному свету. Это различие обусловлено только большей точностью собственно спектрофотометрического измерения. Кроме того, при исследовании растворов или экстрактов окрашенного волокна нет осложнений, связанных с неравномерностью распределения красителя на волокне. Поэтому измерения в отраженном свете применяются главным образом для измерения цвета и сравнения цветовых различий, тогда как количество красителя в волокне лучше определять, переводя краситель в растворитель.[1]
Определение красителей в волокне методами полного растворения
Красители в полимерных материалах, таких, как пленки или текстильные волокна, обычно определяют спектрофотометрически, либо полностью растворяя полимер, содержащий краситель в общем для них растворителе, либо экстрагируя краситель из полимера растворителем, который растворяет только краситель. Первый метод (полного растворения) применим и к таким красителям, которые не удается экстрагировать из волокна, например ковалентно связанные активные красители. Он сравнительно прост, если можно найти подходящий растворитель.
Определение красителя в волокне экстракционными методами
Экстракционные методы применимы к красителям, растворимым в экстрагенте и связанным с волокном за счет ионных и других невалентных взаимодействий. Для превращения красителя в растворимую форму можно сочетать экстракцию с химической реакцией.
Требование к растворителям, применяемым для экстракции, те же, что и к растворителям, растворяющим волокно, но необходимо не растворение волокна, а только его набухание, иначе частицы волокна вызовут мутность раствора и затруднят спектрофотометрическое определение красителя в экстракте.
Экстракция красителя полимера представляет собой процесс, обратный крашению, и является десорбцией неионного красителя из полиэфирного или полиамидного волокна. Диффузия красителя из волокна в растворитель облегчается подвижностью полимерных цепей. Эта подвижность может быть увеличена за счет термической энергии растворителя, который вызывает набухание полимера, делает его более пластичным. Способность расворителя вызывать набухание полимера, необходимое для эффективной экстракции, может быть оценена по параметру растворимости растворителя.
2. ОПИСАНИЕ УСТАНОВОК И ОБРАБОТКИ РЕЗУЛЬТАТОВ ОПРЕДЕЛЕНИЙ
2.1.Установка для прямой фотометрии
Фотометрический метод анализа основан на избирательном поглощении молекулами или ионами определяемого соединения электромагнитного излучения с длиной волны от 400 до 800нм [6]. В основе прямой фотометрии лежит непосредственное измерение оптической плотности серии стандартных растворов определяемого соединения при выбранной длинне волны и нахождении его концентрации одним из методов количественного анализа на основе закона Бугера–Ламберта–Бера [2]. В лабораторной практике для определения величины оптической плотности А используют приборы, называемые колориметрами. Фотоколориметр КФК-2 является однолучевым прибором, предназначенным для измерения оптической плотности в отдельных диапазонах длинн волн от 315 нм до 980нм. Для выделения отдельных участков длинн волн (монохроматизации света) используют встроенные в прибор светофильтры. Принципиальная cхема прибора представлена на рис.1.
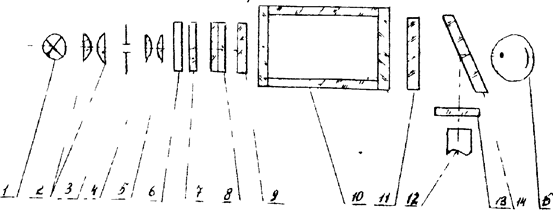
Рис. 1. Принципиальная оптическая схема фотоколорифера КФК-2.
Световой поток от лампы накаливания (1) проходит через систему линз (2,4,5), диафрагму (3), светофильтры (6,7,8) и попадает на кювету с исследуемым раствором (10), который находится между защитным стеклом (9,11). Цветные светофильтры (8) предназначены для выделения узких участков спектра из сплошного спектра излучения лампы (1).
Теплозащитный светофильтр (6) вводится в световой поток при работе в видимой области спектра (400−590 нм), а нейтральный светофильтр (7) используется для ослабления светового потока при работе в спектральном диапазоне 400−540 нм. Фотоприемник (12,15) работает в разных областях спектра: фотоэлемент Ф−26 (15) в области длин волн 315−540 нм; фотодиод ФД−24К (12) в области спектра 590−980 нм.
Пластина (14) делит световой поток на два: 10% светового потоканаправляется на фотодиод и 90% на фотоэлемент.
Для уравнивания фототоков, снимаемых с фотоприемника при работе с различными цветными светофильтрами (8), перед ними установлен светофильтр (13).
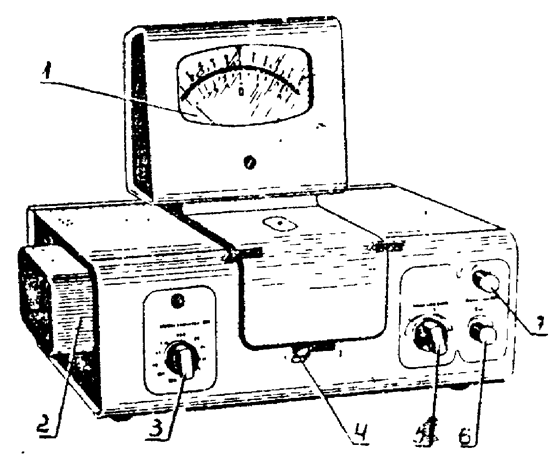 |
Внешний вид прибора КФК−2 представлен на рис.2. Методика проведения измерений на приборе описана в разделе 4.2.2. методического указания[6].
Рис. 2. Внешний вид схема фотоколорифера КФК-2.
1 – шкала коэффициентов пропускания;
3 – ручка для введения необходимого светофильтра;
4 – ручка для введения кювет в световой поток;
5,6,7 – ручки для установления чувствительности прибора.
2.2.Установка для потенциометрического титрования
Метод потенциометрического титрования основан на измерении потенциала индикаторного электрода в процессе титрования и установлении зависимости между его величиной и объемом прибавленного раствора. Выбор индикаторного электрода зависит от природы определяемого иона и типа протекающей реакции.
При кислотно-основном титровании в качестве индикаторного электрода используют стеклянный электрод, на поверхности которого протекает реакция обмена между ионами водорода, находящимися в анализируемом растворе, и ионами натрия, находящимися в стеклянной мембране.
В процессе потенциометрического титрования сначала происходит постепенное изменение потенциала индикаторного электрода, а вблизи точки эквивалентности потенциал резко изменяется и наблюдается его скачок.
Схема установки для потенциометрического титрования с автоматической фиксацией конечной точки титрования и функциональная схемав БАТ-15 представлены на рис. 3.
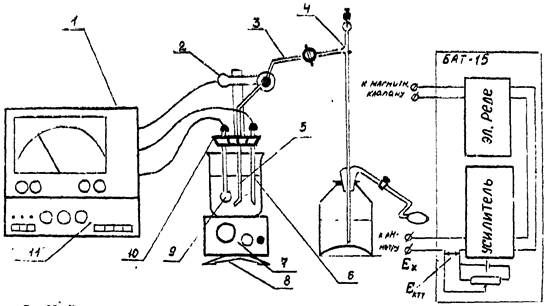
Рис. 3. Схема установки для потенциометрического титрования с автоматической фиксацией КТТ.
1 – рh-метр-милливольтметр;
2 – электромагнитный клапан;
3 – резиновая трубка;стаканчик с анализируемым раствором;
4 – полуавтоматическая бюретка;
5 – дозирующий капилляр;
6 – хлорсеребряный электрод сравнения;
7 - магнитная мешалка;
8 – штатив;
9 - стеклянный индикаторный электрод;
10 – держатель электродов;
11 – БАТ-15.
Методика титрования с БАТ-15 описана в разделе 3.2.2. методического указания [5].
2.3. Установка для высокочастотного титрования
Метод высокочастотного титрования основан на измерении электропроводности раствора при использовании тока высокой частоты (более 0.1 МГц) в процессе титрования для установления конечной точки титрования. Под воздействием электромагнитного поля высокой частоты происходит не только перемещение ионов электролита между электродами, но и поляризация молекул раствора. Электропроводность раствора повышается с увеличением подвижности ионов, концентрации ионов и температуры раствора, поэтому высокочастотное титрование следует проводить при постоянной температуре, титрант должен быть более высокой концентрации, чем титруемый раствор, чтобы его электропроводность не уменьшалась в результате разбавления титрантом. Для высокочастотного титрования используют те химические реакции, при которых наблюдаются различие в подвижности ионов, находящихся в растворе до и после точки эквивалентности [7].
Общий вид установки для высокочастотного титрования (титратор ТВ-6Л1), с помощью которой измеряют силу тока – величину, обратно пропорциональную электропроводности раствора, представлен на рис. 4 [7]
1 – держатель;
2 – бюретка для титрования;
3 – ячейка для высокочастотного титрования;
4 – ручка «грубо» для установления стрелки микроампера на необходимое деление шкалы;
5 – ручка, регулирующая начальное положение стрелки микроамперметра;
6 – ручка для включения прибора;
7 – ручка, необходимая для включения магнитной мешалки;
8 – ручка, регулирующая частоту вращения размешивателя;
9 – переключатель чувствительности прибора.
Рис. 4. Общий вид титратора ТВ-6Л1.
 |
Важным преимуществом высокочастотного титрования является отсутствие контакта анализируемого раствора с электродами, поэтому вместо платиновых могут использоваться электроды из любых металлов,например, более дешевые стальные, которые находятся с наружной стороны электролитической ячейки, внешний вид которой представлен на рис.5.
Рис.5. Ячейка для высокочастотного титрования.
1 – стальные электроды;
2 – электролитическая ячейка.
2.4. Математическая обработка результатов определения
Математическую обработку результатов определения проводили методами математической статистики для малых выборок, используя следующие формулы [2]:
· Среднее арифметическое С = åС i /n,
где С i – единичный результат определения,
n – количество параллельных определений
· Оценка стандартного отклонения единичного определения,
S i = Гå(С i -С)2 /(n-1),
· Относительное стандартное отклонение S r = S i /С,
· Величина, определяющая доверительный интервал δ=S i t табл. / n,
где t табл. – критерий Стьюдента.
· Проверку значимости расхождений между средним и действительным значениями определяли по t-критерию Стьюдента, рассчитанному по формуле tэксп. =|C-a|√n/Si. . Полученное значение сравнивали с табличным t табл. при Р = 0.95 и f = n-1. Если t эксп. <t табл. , то результаты определений можно считать правильными, а расхождение между средним и действительным значениями обусловлено только случайными погрешностями. Если t эксп. > t табл. , то расхождение между средним и действительным значениями значимо и обусловлено случайными и систематическими погрешностями.
· Относительную ошибку определений рассчитывали по формуле
∆отн =|C-a|100% / a
· Расчет коэффициентов регрессии для уравнений градуировочных графиков А=аС+в проводили методом наименьших квадратов по следующим формулам :
а = (n åC i A i - åC i åА i ) / (nå(C i ) 2 – (åC i )2 ),
в = (å(C i )2 åA i - åC i å C i А i ) / (nå(C i ) 2 – (åC i )2 )
· Коэффициент коррелляции рассчитывается по формуле:
r = (n åC i A i - åC i åА i ) / {(n å(C i ) 2 - (åC i )2 ) (nå(А i 2 - (åА i )2 }1/2 .
3. РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ.
3.1. Исследование условий фотометрического определения красителя в ванне крашения кислотными красителями.
3.1.1. Выбор светофильтра .
Для выбора светофильтра в одну кювету заливали анализируемый раствор ванны, а в другую – кювету сравнения – дистиллированную воду и измеряли оптическую плотность раствора ванны А со всеми светофильтрами. Результаты измерений зафиксированны в табл. 1.
ТАБЛ. 1 .
| l ,нм |
315 |
364 |
400 |
440 |
490 |
540 |
590 |
670 |
750 |
870 |
980 |
| Ак.к. |
- |
0.190 |
0.180 |
0.220 |
0.500 |
0.270 |
0.020 |
0.020 |
- |
- |
- |
| Ак.з. |
- |
0.235 |
0.195 |
0.105 |
0.252 |
0.220 |
0.435 |
0.305 |
0.245 |
- |
- |
По результатам, представленным в табл. 1, построили зависимость оптической плотности А от длины волны л для кислотного красного (1) и кислотного антрахинонового ярко-зеленого (2) (рис. 6).
Рис. 6. Спектры поглощения кислотного красного (1) и кислотного антрахинонового (2) красителей.
Как видно из табл. 1 и рис. 6, определение в ванне крашения кислотным красным необходимо проводить при длине волны, соответствующей максимуму поглощения анализируемого раствора, т. е. l=490нм , а кислотным антрахиноновым ярко-зеленым при l=590 нм.
3.1.2. Построение градуировочного графика, установление интервала его линейности и стабильности во времени
Для приготовления серии стандартных растворов необходимо взять 5 мерных колб вместимостью 50.00 мл, в каждую из которых поместить соответственно 1.00, 2.00, 3.00, 4.00 и 5.00 мл раствора кислотной ванны крашения с известной концентрацией и довести до метки дистиллированной водой.
Расчет концентрации красителя в каждой колбе проводится по формуле C i = Cкр-ля Vi /Vк ,
где Скр-ля – известная концентрация красителя (для кислотного красного – 0.2440 г/л; для кислотного антрахинонового ярко-зеленого - 0.2240г/л),
Vi – объем пробы красителя, мл.
Vк – объем мерной колбы, мл.
Затем измеряется оптическая плотность (А) полученных стандартных растворов поочередно при выбранном свтофильтре (см. раздел 3.1.1.).
Аналогичные измерения проводятся спустя неделю для проверки стабильности градуировочного графика во времени. Используя метод наименьших квадратов строим градуировочный график.
Данные для построения градуировочного графика для кислотного красного представлены в табл. 2, а градуировочный график - на рис.7.
ТАБЛ. 2 .
| С i , г /л |
0.0048 |
0.0097 |
0.0146 |
0.0195 |
0.0244 |
|
| А 1 |
11.09.2002г. |
0.175 |
0.300 |
0.500 |
0.610 |
0.790 |
| А 2 |
18.09.2002г. |
0.170 |
0.290 |
0.430 |
0.600 |
0.750 |
Рис 7. Градуировочный график для кислотного красного
Данные для построения градуировочного графика кислотного антрахинонового ярко-зеленого представлены в табл. 3. Градуировочный график представлен на рис. 8.
ТАБЛ. 3.
| С i , г /л |
0.0045 |
0.0090 |
0.0134 |
0.0179 |
0.0224 |
|
| А 1 |
11.09.2002г. |
0.025 |
0.075 |
0.125 |
0.155 |
0.238 |
| А 2 |
18.09.2002г. |
0.049 |
0.973 |
0.135 |
0.171 |
0.218 |
Рис. 8. Градуировочный график для кислотного антрахинонового ярко-зеленого
Как видно из рис. 7, градуировочный график кислотного красного линеен в интервале концентраций от 0.0048 г/л до 0.0244 г/л и стабилен во времени.
Анализируя рис. 8, делаем вывод, что график кислотного антрахинонового ярко-зеленого линеен в интервале концентраций от 0.0045 г/л до 0.0224 г/л и стабилен во времени.
Были рассчитаны коэффициенты для уравнений градуировочных графиков: А = аС + в.
Для кислотного красного: а=39,828,
в=0.00023,
r=0.9910.
А = 36.83С + 0.00023.
Для кислотного антрахинонового ярко-зеленого: а=9.5900,
в=0.0007,
r=0.9907.
А = 9.59С = 0.0007.
Коэффициенты коррелляции r больше 0.99. Следовательно, полученные градуировочные графики можно использовать для количественных определений.
3.1.3. Расчет разбавления пробы
Концентрация красителя модельного раствора красильной ванны приближенно составляет С ~ 0.2 г/л. При определении кислотного красного середине градуировочного графика соответствует концентрация Скр.= 0.012г/л. Чтобы при разбавлении модельного раствора ванны крашения кислотного красного попасть на середину градуировочного графика, пробу необходимо разбавить в 20 раз ( С / Cкр.~20).
При определении кислотного антрахинонового ярко-зеленого середина градуировочного графика приходится на Сзел.=0.011 г/л. Т. к. концентрация в модельном растворе ванны крашения кислотного антрахинонового ярко-зеленого С ~ 0.2, то чтобы попасть на середину градуировочного графика, пробу необходимо разбавить в 10 раз.
3.1.4. Проверка правильности результатов определения
Проверку правильности результатов определения проводим путем анализа модельных растворов красильной ванны с известными концентрациями: акрасн. = 0.2293 г/л; азелен. = 0.1864 г/л.
Для разбавления модельного раствора ванны крашения кислотного красного в 20 раз в колбу на 100 мл помещаем 5.00 мл анализируемого раствора, доводим до метки достиллированной водой и проводим измерения оптической плотности раствора при выбранном светофильтре (см. раздел 3.1.1). Параллельные определения проводим 3. Полученные данные представлены в табл.4.
Табл. 4.
| № измер-я |
А |
С фотометрир., г /л |
С анализир.,г /л |
| 1 |
0.415 |
0.0114 |
0.228 |
| 2 |
0.412 |
0.0113 |
0.226 |
| 3 |
0.407 |
0.0112 |
0.224 |
Математическая обработка результатов определения для модельного раствора ванны крашения кислотного красного представлена в табл.4.1.
ТАБЛ. 4.1
| С, г /л |
n |
Sr |
d , г /л |
С- d…….С+ d |
tтабл. |
tэксп. |
| 0.226 |
3 |
0.0087 |
0.005 |
0.221….0.231 |
4.303 |
3.350 |
Поскольку относительная ошибка опрелелений составляет 1.3.%, а tэксп. <tтабл. (3.35<4.303), то результат определения считается правильным, а расхождение между средним и действительным значениями обусловлено только случайными погрешностями.
Для разбавления модельного раствора ванны крашения кислотного антрахинонового ярко-зеленого в 10 раз в колбу на 50.00 мл помещаем 5.00 мл анализируемого раствора, доливаем до метки дистиллированной водой и измеряем оптическую плотность полученного раствора. Проводим 4 параллельных определения. Полученные данные представлены в табл. 5.
ТАБЛ. 5.
| № измер-я |
А |
С фотометрир., г /л |
С анализир., г /л |
| 1. |
0.175 |
0.0183 |
0.183 |
| 2. |
0.173 |
0.0173 |
0.173 |
| 3. |
0.168 |
0.0176 |
0.176 |
| 4. |
0.182 |
0.0197 |
0.197 |
Математическая обработка результатов определения для модельного раствора красильной ванны кислотного антрахинонового ярко-зеленого представлена в табл. 5.1.
ТАБЛ. 5.1
| С, г /л |
n |
Sr |
d , г /л |
С- d…….С+ d |
tтабл. |
tэксп. |
| 0.182 |
4 |
0.045 |
0.015 |
0.167….0.197 |
3.182 |
2.315 |
Т. к. относительная ошибка определений составляет 2.23%, а tэксп. < tтабл. (2.315<3.182), результат определения может считаться правильным, а расхождение между средним и действительным значениями обусловлено только случайными погрешностями.
3.2. Определение концентраций красителей в технологических растворах красильных ванн методом прямой фотометрии.
По методике, описанной в разделе 3.1, были проанализированны технологические ванны крашения кислотного красного и кислотного антрахинонового ярко-зеленого. Полученные данные представлены в табл. 6 и 7. Математическая обработка результатов определения кислотного красного и кислотного антрахинонового ярко-зеленого в технологических ваннах представлена в табл. 6.1 и 7.1. соответственно.
Результаты определения концентрации кислотного красного в технологической ванне крашения
ТАБЛ. 6.
| № измер-я |
А |
С фотометрир. г /л |
С анализир.,г /л |
| 1. |
0.350 |
0.096 |
0.192 |
| 2. |
0.320 |
0.088 |
0.176 |
| 3. |
0.340 |
0.093 |
0.186 |
| 4. |
0.330 |
0.090 |
0.181 |
ТАБЛ. 6.1.
| С, г /л |
n |
Sr |
d , г /л |
С- d…….С+ d (Р=0.95) |
| 0.184 |
4 |
0.045 |
0.013 |
0.171….0.197 |
Результаты определения концентрации кислотного антрахинонового ярко-зеленого в технологической ванне крашения
ТАБЛ. 7
| № измер-я |
А |
С фотометрир., г /л |
С анализир., г /л |
| 1. |
0.156 |
0.0163 |
0.163 |
| 2. |
0.161 |
0.0168 |
0.168 |
| 3. |
0.162 |
0.0170 |
0.170 |
| 4. |
0.168 |
0.0171 |
0.171 |
ТАБЛ. 7.1.
| С, г /л |
n |
Sr |
d , г /л |
С- d…….С+ d (Р=0.95) |
| 0.168 |
4 |
0.037 |
0.011 |
0.157….0.179 |
3.3. Иследование условий определения концентрации уксусной кислоты в ваннах крашения кислотными красителями методом полуавтоматического потенциометрического титрования.
В основе определения концентрации уксусной кислоты в кислотных красильных ваннах лежит следующая реакция:
СН3 СООН + NaОН - CН3 СОО Na + Н2 О.
3.3.1. Расчет объема пробы уксусной кислоты
Расчет объема пробы уксусной кислоты проводят по закону эквивалентности: C пр. Vпр. = Cтитр. Vтитр. ,
где Vтитр. и Vпр. - объем титранта (гидроксида натрия) и объем пробы анализируемого раствора соответственно, мл.
Ститр и Спр. – концентрация титранта и концентрация уксусной кислоты в анализируемом растворе красильной ванны соответственно, моль/л.
Концентрация СН3 СООН в модельных растворах красильных ванн ~ 0.3 г/л, что составляет 0.005 моль/л, а концентрация NaОН ~ 0.09 моль/л. Задаемся объемом титранта в конечной точке титрования = 1.5 мл, тогда Cпр. = (Cтитр х Vтитр. . )/ Vпр. = (0.09моль/л х 1.5мл) /0.005моль/л = 30.00 мл.
Выбираем объем пробы =25.00 мл.
3.3.2 Выбор Ектт для полуавтоматического потенциометрического титрования
В стаканчик для титрования помещаем с помощью пипетки 25.00 мл аналилируемого модельного раствора кислотной ванны крашения. Затем ставим стаканчик на магнитную мешалку, опускаем в него электроды так, чтобы они находились в растворе и не соприкасались со стенками стаканчика, помещаем в полученный раствор размешиватель и включаем мешалку. Далее прибавляем из бюретки раствор титранта с известной концентрацией (СNaOH =0.08800моль/л) по 0.20 мл, а вблизи точки эквивалентности по 0.10 мл для более точного построения кривых титрования. После каждого прибавления титранта измеряем изменение значения потенциала индикаторного электрода и фиксируем полученные результаты. Проводим 3 параллельных определения.
По полученным данным строим кривые потенциометрического титрования в координатах “потенциал электрода, Е,мВ – объем титранта V,мл”, пример одной из которых представлен на рис. 9.
Рис. 9. Кривая потенциометрического титрования.
Как видно из рис.9, потенциал в конечной точке титрования Ектт = -100 мВ. Во избежании перетитровывания на БАТ-15 устанавливаем меньший потенциал: Ектт = -15мВ. Выбранные условия для полуавтоматического потенциометрического титрования представлены в табл. 8.
ТАБЛ. 8.
| Ектт, мВ |
Время выдержки,с |
Диапазон титрования |
Зона импульсной подачи титранта, мВ |
| -15 |
15 |
узкий |
200 |
3.3.3 Проверка правильности результатов определения
Проверку правильности результатов определения концентрации уксусной кислоты в ваннах крашения кислотными красителями проводили путем анализа модельных растворов красильных ванн с известной концентрацией уксусной кислоты: для кислотного красного действительное значение ак.к =0.472 г/л , для кислотного антрахинонового ярко-зеленого ак.з. =0.528г/л.
Дальнейшие определения проводим автоматически, задав на БАТ-15 необходимые условия, представленные в табл. 8. Для этого отбираем пипеткой 25.00 мл анализируемого раствора модельной ванны крашения и помещаем его в стаканчик для титрования. Ставим стаканчик на магнитную мешалку, помещаем в полученный раствор размешиватель, опускаем электроды, включаем мешалку и автоматическую подачу титранта. После окончания титрования (сигналом является загорающаяся лампочка «конец») фиксируем объем титранта, пошедшего на титрование анализируемого модельного раствора. Концентрацию уксусной кислоты рассчитываем по формуле: С CH 3 COOH = С NaOH V КТТ M CH 3 COOH /V пр.
Результаты определения концентрации уксусной кислоты в модельных ваннах крашения кислотным красным представлены в табл. 9, а кислотным антрахиноновым ярко-зеленым в табл.10.
Табл.. 9
| V КТТ , мл |
2.31 |
2.25 |
2.27 |
| C CH 3 COOH ,г/л |
0.488 |
0.476 |
0.480 |
Табл.10
| V КТТ , мл |
2.53 |
2.49 |
2.52 |
2.50 |
| C СH 3 COOH ,г/л |
0.534 |
0.527 |
0.532 |
0.528 |
Была проведена математическая обработка результатов определения концентрации уксусной кислоты в модельных ваннах крашения кислотными красителями, результаты которой представлены в табл. 9.1 и 10. 1 соответственно.
Табл. 9.1.
| С, г /л |
n |
Sr |
δ, г /л |
С-δ…….С+δ (Р=0.95) |
| 0.481 |
3 |
0.013 |
0.015 |
0.466….0.496 |
Т. к. относительная ошибка определений составляет 2.2%, а tэксп. < tтабл. (2.315<3.182), результат определения может считаться правильным, а расхождение между средним и действительным значениями обусловлено только случайными погрешностями
Табл. 10.1.
| С, г /л |
n |
Sr |
δ, г /л |
С-δ…….С+δ (Р=0.95) |
| 0.530 |
4 |
0.006 |
0.005 |
0.525….0.535 |
Поскольку относительная ошибка определений составляет 0.38%, а tэксп. < tтабл. (2.403<3.182), результат определения может считаться правильным, а расхождение между средним и действительным значениями обусловлено только случайными погрешностями.
3.4. Определения концентрации уксусной кислоты в технологических ваннах крашения кислотными красителями методом полуавтоматического потенциометрического титрования.
Методика определения концентрации уксусной кислоты в красильной ванне описана в разделе 3.3.3. Результаты определений концентраций уксусной кислоты в технологических ваннах крашения кислотными красителями представлены в табл. 11 для кислотного красного и табл. 12 для кислотного антрахинонового ярко-зеленого. Результаты математической обработки полученных данных в табл. 11.1 и 12.1 соответственно.
Табл.11
| V КТТ , мл |
1.71 |
1.68 |
1.64 |
1.72 |
| C СH 3 COOH ,г/л |
0.301 |
0.296 |
0.290 |
0.302 |
Табл.11.1
| С, г /л |
n |
Sr |
δ, г /л |
С-δ…….С+δ (Р=0.95) |
| 0.297 |
4 |
0.019 |
0.009 |
0.288….0.306 |
Табл.12.
| V КТТ , мл |
3.15 |
3.18 |
3.20 |
3.16 |
| C СH 3 COOH ,г/л |
0.666 |
0.672 |
0.675 |
0.668 |
Табл.12.1
| С, г /л |
n |
Sr |
δ, г /л |
С-δ…….С+δ (Р=0.95) |
| 0.670 |
4 |
0.006 |
0.006 |
0.664….0.676 |
3.5 Исследование условий определения концентрации сульфата натрия в ваннах крашения кислотными красителями методом высокочастотного титрования.
В основе определения концентрации сульфата натрия в ваннах крашения кислотными красителями лежит следующая реакция:
Na2 SO4 + Ba(CH3 COO) 2 = 2 CH3 COONa + BaSO4
3.5.1. Расчет объема пробы
Расчет объема пробы проводим по закону эквивалентности
C пр. Vпр. = Cтитр. Vтитр. ,
где Cпр. и Cтитр. = концентрация сульфата натрия в анализируемом растворе и концентрация ацетата бария соответственно, моль/л
Vпр. и Vтитр. - объем пробы анализируемого раствора и объем ацетата бария в конечной точке титрования соотвотственно,мл.
Концентрация ацетата бария составляет ~ 0.25 моль/л.Задаемся объемом титранта в конечной точке титрования =5.00 мл. Концентрация сульфата натрия в модельной ванне крашения кислотным красным составляет ~ 0.05 моль/л. Тогда Vпр =(0.25 5.00)/0.05=25.0 мл. Выбираем объем пробы 25.00 мл.
Для кислотного антрахинонового ярко-зеленого концентрация сульфата натрия ~ 0.07 моль/л. Поэтому Vпр =(0.25 5.00)/0.07=17.8мл. Выбираем объем пробы = 20.00 мл.
3.5.2.Установление концентрации титранта
Концентрацию ацетата бария устанавливают путем титрования. Для этого в стаканчик для титрования наливают из бюретки 25.00 мл раствора серной кислоты с известной концентрацией С(H2 SO4 )=0.05000 моль/л, доливают дистиллированную воду до уровня 2-3мм выше уровня электродов, помещают в анализируемый раствор размешиватель. В ячейку для титрования вставляют стакан, ручкой “грубо” устанавливают стрелку микроамперметра в положение “0”, ставят чувствительность прибора на “4”, включают магнитную мешалку, регулируя частоту вращения размешивателя таким образом, чтобы глубина образующейся на поверхности раствора воронки была не менее 3 мм, но не опускалась ниже уровня электрода. Бюретку для титрования заполняют раствором ацетата бария и начинают титровать, добавляя по 0.50 мл титранта (а в близи точки эквивалентности по 0.20 мл для более точного построения кривых титрования), и фиксируя изменения показаний прибора в процессе титрования. Проводят 4 параллельных определения. По результатам титрования строят кривые титрования в координатах “сила тока, мкА – объем титранта, мл”, пример одной из которых представлен на рис. 10.
Рис. 10. Кривая потенциометрического титрования Ba(CH3 COO) 2 стандартным раствором Н2 SO4
Объем титранта в конечной точке титрования определяют путем нахождения точки пересечения линейных участков кривой титрования. Как видно из рис. 10, среднее значение Vктт =5.02мл.
Молярную концентрацию титранта рассчитывают на основе закона эквивалентности по формуле: С Ва(CH 3 COO) 2 = С NaOH V пр /V КТТ
Где С H2 SO4 – концентрация стандартного расвора серной кислоты, моль/л;
Vпр и Vктт - объем пробы стандартного раствора Н2 SO4 раствора и объем титранта в конечной точке титрования соответственно, мл.
С Ва(CH 3 COO) 2 = (0.0500 х 25, 00)/5.020 = 0.02488моль/л.
3.5.3. Проверка правильности результатов определений
Проверку правильности результатов определений проводят путем анализа модельных ванн крашения кислотными красителями с известными концентрациями содержащегося в них сульфата натрия. Для для кислотного красного действительное значение ак.к =7.350 г/л, а для кислотного антрахинонового ярко-зеленого ак.з. =10.03 г/л.
Анализируемый раствор модельной ванны крашения кислотным красным отбирают с помощью пипетки в объеме 25.00 мл (для кислотного антрахинонового ярко-зеленого в объеме 20.00 мл) и помещают в стаканчик для титрования, далее проводят титрование аналогично описанному в разделе 3.5.2., при этом стрелку микроамперметра устанавливают в положение «50» во избежании зашкаливания, а чувствительность прибора повышают до «2». Проводят 3 параллельных определения. По результатам титрования строят кривые титрования, пример одной из которых представлен на рис.11. по полученным кривым определяем объем титранта, пошедшего на титрование анализируемого модельного раствора.
Концентрацию сульфата натрия рассчитываем по формуле:
С Na2 SO4 . =С Ва(CH 3 COO) 2 М Na2 SO4 Vктт /Vпр .
Результаты определения концентрации сульфата натрия в модельных ваннах крашения кислотным красным представлены в табл. 11, а кислотным антрахиноновым ярко-зеленым в табл.12.
Табл.. 11
| V КТТ , мл |
5.25 |
5.30 |
2.40 |
| C Nа 2 SO4 ,г/л |
7.419 |
7.489 |
7.631 |
Табл.12
| V КТТ , мл |
5.70 |
5.80 |
5.50 |
| C Nа 2 SO4 ,г/л |
10.14 |
10.07 |
9.98 |
Была проведена математическая обработка результатов определения концентрации сульфата натрия в модельных ваннах крашения кислотными красителями, результаты которой представлены в табл. 11.1 и 12. 1 соответственно.
Табл. 11.1.
| С, г /л |
n |
Sr |
δ, г /л |
С-δ…….С+δ (Р=0.95) |
| 7.51 |
3 |
0.014 |
0.27 |
7.24….7.78 |
Т. к. относительная ошибка определений составляет 2.18%, а tэксп. < tтабл. (2.566<4.303), результат определения может считаться правильным, а расхождение между средним и действительным значениями обусловлено только случайными погрешностями
Табл. 12.1.
| С, г /л |
n |
Sr |
δ, г /л |
С-δ…….С+δ (Р=0.95) |
| 10.06 |
3 |
0.0065 |
0.14 |
9.92….10.20 |
Поскольку относительная ошибка определений составляет 0.30%, а tэксп. < tтабл. (2.103<4.303), результат определения может считаться правильным, а расхождение между средним и действительным значениями обусловлено только случайными погрешностями.
3.6. Определения концентрации сульфата натрия в технологических ваннах крашения кислотными красителями методом высокочастотного титрования.
Методика определения концентрации сульфата натрия описана в разделе 3.5.3. Результаты определений сульфата натрия в технологической красильной ванне кислотного красного и их математическая обработка представлены в табл. 13 и табл. 13.1.соответственно.
Результаты определений сульфата натрия в технологической к ванне крашения кислотным антрахиноновым ярко-зеленым и их математическая обработка представлены в табл. 14 и табл. 14.1. соответственно.
Табл.. 13
| V КТТ , мл |
4.50 |
4.40 |
4.60 |
| C Nа 2 SO4 ,г/л |
6.37 |
6.42 |
6.28 |
Табл. 13.1.
| С, г /л |
n |
Sr |
δ, г /л |
С-δ…….С+δ (Р=0.95) |
| 6.36 |
3 |
0.011 |
0.18 |
6.18….6.54 |
Табл.. 14.
| V КТТ , мл |
6.58 |
6.65 |
6.62 |
| C Nа 2 SO4 ,г/л |
11.62 |
11.74 |
11.69 |
Табл. 14.1.
| С, г /л |
n |
Sr |
δ, г /л |
С-δ…….С+δ (Р=0.95) |
| 11.68 |
3 |
0.005 |
0.16 |
11.52…..11.84 |
ВЫВОДЫ
1. Исследование условий определения кислотных красителей в ваннах крашения методом прямой фотометрии показало, что
- определение кислотного красного красителя необходимо проводить со светофильтром, имеющим λmax =490нм, а кислотного антрахинонового ярко-зеленого со светофильтром при λmax =590 нм;
- градуировочный график для кислотного красного линеен в интервале концентраций от 0.0048 до 0.0244 г/л и стабилен во времени, градуировочный график для кислотного антрахинонового ярко-зеленого линеен в интервале концентраций от 0.0045 до 0.0224 г/л и также стабилен во времени;
- при определении концентрации кислотного красного и кислотного антрахинонового ярко-зеленого ~ 0.2 г/л пробы необходимо разбавить в 20 и 10 раз соответственно;
- Проверка правильности результатов определений показала отсутствие систематических погрешностей; относительная ошибка определений Δотн = 1.31% при определении кислотного красного и Δотн =2.23% при определении кислотного антрахинонового ярко-зеленого.
- При определении красителей воспроизводимость можно считать удовлетворительной, т. к. относительное стандартное отклонение Sr <0.0045
2. Исследование условий определения содержания уксусной кислоты в ваннах крашения кислотными красителями методом полуавтоматического потенциометрического титрования показало, что
- при концентрации титранта (раствора гидроксида натрия)~ 0.9моль/л объем пробы титруемого раствора красильной ванны должен составлять 25.00 мл при содержании уксусной кислоты в интервале концентраций от 0.4 до 0.7 г/л;
- автоматическое титрование необходимо проводить до Е=-15 мВ;
- проверка правильности результатов определений показала отсутствие систематических погрешностей, относительная ошибка определений Δотн =2.17% при определении кислотного красного и Δотн =0.38% при определении кислотного антрахинонового ярко-зеленого.
- воспроизводимость результатов определений можно считать хорошей, т. к. относительное стандартное отклонение Sr =0.026
3. Исследование условий определения содержания сульфата натрия в ваннах крашения кислотными красителями методом высокочастотного титрования показало, что:
- объем пробы титруемого раствора при концентрации тиранта С[Ва(CH3 COO)2 ] ~ 0.25 г/л составляет 25.00 мл;
- проверка правильности результатов определений показала отсутствие систематических погрешностей; относительная ошибка определения Δотн =2.18% при определении кислотного красного и Δотн =0.31% при определении кислотного антрахинонового ярко-зеленого.
- воспроизводимость результатов определений можно считать хорошей, т. к. относительное стандартное отклонение Sr ≤0.014.
СПИСОК ЛИТЕРАТУРЫ .
1. Булушева Н.Е., Лабораторный практикум по химическим технологиям волокнистых материалов, М.,1986.
2. Быкова Л.Н., Новиков А.В., Чеснокова О.Я., Аналитическая химия, М., МГТУ им. Косыгина,2002.
3. Кричевский Г.Е, Корчагин Н.В., Химические технологии текстильных материалов, М., 1985.
4. Степанов Б.И., Введение в химию и технологию органических красителей, М., Химия, 1977.
5. Методическая разработка к лабораторным работам по курсу “Аналитическая химия”, часть 1 “Электрохимические методы анализа”, М., РИО МГТА,1993.
6. Методическая разработка к лабораторным работам по курсу “Аналитическая химия”, раздел “Контроль загрязнений окружающей среды” под редакцией Быковой Л.Н.. М., РИО МГТА,1992.
7. Методическая разработка к лабораторным работам по курсу “Аналитическая химия”, раздел “Титриметрические методы анализа с физико-химическими способами установления конечной точки титрования” под редакцией Быковой Л.Н.. М., РИО МГТА ,1993.