| Скачать .docx |
Курсовая работа: Акридон. Его получение, свойства и применение
“СИНТЕЗ АКРИДОНА”
Содержание
1. Введение________________________________________________ 3
1.1 Свойства акридона______________________________________ 3
1.2 Применение акридона___________________________________ 3
1.3 Синтезы акридона_______________________________________ 4
2. Обзор литературы________________________________________ 5
2.1 Реакции замыкания цикла. Типы реакций___________________ 6
2.2 Замещение при насыщенном атоме углерода________________ 8
2.3 Внутримолекулярное нуклеофильное присоединение к карбонильной группе_____________________________________________ 10
2.4 Внутримолекулярное нуклеофильное присоединение к другим двойным связям__________________________________________________ 11
2.5 Электролитические реакции_____________________________ 13
2.6 Акридин______________________________________________ 16
3. Обсуждение результатов_________________________________ 20
4. Экспериментальная часть_________________________________ 20
4.1 Реагенты и оборудование______________________________________ 20
4.2 Методика эксперимента_______________________________________ 21
5. Выводы________________________________________________ 21
6. Библиографический список_______________________________ 22
ВВЕДЕНИЕ
1.1 Свойства акридона. Акридон (9-акридон, 9-гидроксиакридин) – очень устойчивое в обычных растворителях желтое вещество игольчатой структуры; оно плавится при высокой температуре (354ο, испр.). Нерастворимо в воде, очень трудно растворимо в этаноле и эфире, хорошо растворимо в горячей уксусной кислоте. Акридон отличается от изомерных ему оксиакридинов отсутствием явно выраженных кислых и основных свойств. Спектр акридона тоже значительно отличается от спектров оксиакридинов. Вопрос о том, какая формула, кетонная или оксиакридиновая, более точно отражает его свойства, был предметом длительной дискуссии. Если представить себе, что соединение ионизируется, хотя бы в незначительной степени, так что атом кислорода становится анионом, а к атому азота присоединяется освобождающийся водород, то вместо формулы I возникает II (рис. 1).
Молекулярный вес акридона ![]() . Определен криоскопически в феноле; оказалось, что в этих условиях акридон мономерен; однако Xунтер показал, что вещества такого типа могут состоять из коротких цепей молекул, соединенных водородной связью. Идентификация акридонов производится переведением их в соответствующие 5-(n-диэтиламино) фенилакридины.
. Определен криоскопически в феноле; оказалось, что в этих условиях акридон мономерен; однако Xунтер показал, что вещества такого типа могут состоять из коротких цепей молекул, соединенных водородной связью. Идентификация акридонов производится переведением их в соответствующие 5-(n-диэтиламино) фенилакридины.
Не только нитроакридоны, но и некоторые из аминоакридонов (особенно 3-аминоакридон) обнаруживают явные кислые свойства, не характерные для самого акридона; 4-метоксиакридон (единственный из изомеров) является основанием, более сильным, чем акридон, хлоргидрат которого гидролизуется даже в 3 н. соляной кислоте. Причины этого явления до сих пор неясны.
1.2 Применение акридона. Активное изучение обнаружило ряд уникальных свойств, которыми обладают производные акридона. На настоящий момент многие изученные производные акридона обнаружили совсем неожиданное применение – в медицине. Например, акридонуксусная кислота, в принципе известная и ранее. Именно на ее базе была получена активная субстанция, обладающая способностью повышать сопротивляемость организма через индукцию, или стимуляцию эндогенного (внутреннего) интерферона – интерферона, имеющегося в клетках и тканях человека, определяющего устойчивость организма против внутриклеточных паразитов, в частности, вирусов. В результате активных исследований было получено достаточно эффективное вещество, соль М-метил-К-(L,D-глюкопиранозил) аммония 10-метиленкарбоксилат-9-акридона., названная циклофероном (торговое название) — новое, оригинальное и нигде не зарегистрированное. Оно обладает интерферогенной активностью в отношении всех типов интерферона. Исследования показали, что циклоферон обладает не только антивирусными и иммуномоделирующими свойствами, но и способен подавлять развитие ряда микробов [1]. В их числе – возбудители туляремии, бруцеллеза, хламидийных инфекций и др. Существенно помогает циклоферон при лечении злокачественных опухолей. Замечено ингибирующее действие лекарства на развитие инфекции ВИЧ. Проведены испытания препарата, которые показали существенное улучшение состояния больных СПИДом с разными сроками заболевания [2].
Некоторые кубовые красители содержат одновременно антрахиноновые и акридоновые структурные звенья; получаются они из антрахиноновых аналогов фенилантраниловой кислоты. Примерами таких красок служат индантреновый красный и индантреновый фиолетовый; соединения этого типа ценны тем, что они имеют красноватый цвет, а антрахиноновые краски не дают такого оттенка.
1.3 Синтезы акридона. Как правило, акридоны проще всего получать из замещенных дифениламин-2-карбоновых кислот (рис. 2, III), которые кипятят с 6 объемами хлорокиси фосфора до растворения осадка и 30 мин. После растворения. Хлорокись фосфора отгоняют и продукт реакции (соль соответствующего 5-хлоракридина) кипятят в течение 1 часа с 0,5 н. соляной кислотой.
Если исходная дифениламин-2-карбоновая кислота замещена в положении 3′, то образуется смесь двух акридонов с заместителями в положении 4 или 2. Нитро- и метильная группы благоприятствуют образованию 4-производных, а фтор, метокси- и особенно аминогруппа – образованию 2-производных. Относительно влияния хлора мнения расходятся.
Прежде в синтезе акридонов чаще применялась серная кислота. По сравнению с хлорокисью фосфора она удобнее чем, что позволяет избежать стадию образования хлоракридина, но она неактивна в случае нитрозамещенных, а иногда вызывает сульфирование. По-видимому, серной кислотой лучше пользоваться для получения аминоакридонов и самого акридона.
Из акридинов акридон получается с 40%-ным выходом при стоянии смеси 2-нитробензальдегида, бензола, нитрита натрия и концентрированной серной кислоты в течение 5 дней при комнатной температуре. Эта реакция известна с 1909 г., но механизм ее, принятый в настоящее время, был выяснен в 1930г. в результате оживленной дискуссии. Сейчас уже почти не вызывает сомнения, что сначала альдегид и бензол конденсируются в 2-нитробензгидрол, который затем восста-навливается в фенилантранил (рис. 2, IV); последний, как показал Бамбергер, под действием азотистой кислоты каталитически изомеризуется в акридон (рис. 2, V).
N-Замещенные акридоны получаются при нагревании калиевой соли акридона (образуются в спиртовом растворе), например с диметилсульфатом при 100°. Другой метод состоит в нагревании 5-хлоракридина с обычными алкилирующими агентами; реакционную смесь выливают в водный раствор щелочи. Описан метод, согласно которому едкий натр добавляют к раствору соединения, полученного в результате алкилирования (например, к раствору хлористого N-метилакридиния), и осадок (5-окси-N-метилакридан) окисляют хромовым ангидридом.
Акридон сульфируется и нитруется в положения 3 и 3,7, а при бромировании дает 2,3-дибромпроизводное.
Мною апробирован метод получения акридона из фенилантраниловой кислоты. Выбранная реакция принадлежит к реакциям замыкания цикла.
ОБЗОР ЛИТЕРАТУРЫ
Реакции замыкания цикла. Типы реакций.
Реакции замыкания цикла включают внутримолекулярное образование σ-связи. В гораздо большей степени распространены процессы, в которых нуклеофильный центр атакует электрофильный. Среди реакций этого типа можно перечислить следующие: нуклеофильное замещение при насыщенном атоме углерода, нуклеофильное присоединение к ненасыщенному атому углерода и нуклеофильное присоединение – элиминирование. Гетероциклические системы можно также получить в результате внутримолекулярного радикального процесса, электроциклического замыкания цикла с участием сопряженной π-электронной системы или с участием карбенов и нитренов.
Хотя реакция замыкания цикла включает образование одной связи, обычно интермедиат получают из двух или более простых реагентов. Например – синтез пирролов по _ени – Кнорру из 1,4-дикарбонильных соединений и первичных аминов (рис. 3). Стадия циклизации включает нуклеофильную атаку иминного атома азота по карбонильной группе в интермедиате 1.
Следует отметить, что 1,4-дикетон в данном случае дважды выступает в роли электрофила – при взаимодействии с амином и с имином. И в большинстве других синтезов первоначально нуклеофил–электрофильное взаимодействие двух реагентов влечет за собой процесс аналогичного типа, приводящий к замыканию цикла. Различные типы таких взаимодействий показаны ниже.
Примеры компонентов, часто используемых при синтезе гетероциклов, приведены на рис. 4. Соединения, содержащие карбонильную группу (альдегиды, кетоны, хлороангидриды, эфиры карбоновых кислот, а также другие соединения) широко используются как электрофилы.
Реагенты с двумя электрофильными центрами

Реагенты с двумя нуклеофильными центрами

Реагенты с электрофильным и нуклеофильным центрами

Рис. 4. Примеры компонентов различного типа, используемых при синтезе гетероциклов.
Наиболее важный метод синтеза бензоконденсированных гетероциклов состоит в аннелировании гетероциклического кольца к бензольному. При этом существует два основных стратегических подхода: использование диорто-производных бензола и монозамещенных бензолов, в которых opmoположения реагируют как нуклеофилы (т.е. подвергаются нуклеофильной атаке). Например, хинолиновая система может быть получена из 2-аминобензальдегида с использованием двухуглеродного реагента с электрофильным и нуклеофильным центрами. Альтернативный подход основан на использовании анилина и трехуглеродного реагента с двумя электрофильными центрами, например α,β-ненасыщенного кетона. Недостаток второго подхода связан с неоднозначностью протекания процесса при наличии в анилине неэквивалентных орто-положений.
Система номенклатуры для описания возможных типов циклизаций проиллюстрирована на рис. 5. Она определяется характером гибридизации атома, атакуемого нуклеофилом, и тем, происходит ли сдвиг электронов от нуклеофильного центра к эндоциклическому (эндо) или экзоциклическому (экзо) атому.

Рис. 5. Варианты замыкания цикла при нуклеофильно-электрофильном взаимодействии.
Внутримолекулярное замещение при насыщенном атоме углерода – пример экзотет-процесса, а нуклеофильное присоединение к карбонильной группе и процессы присоединения–элиминирования с участием карбонильной группы относятся к экзотриг-типу.
Для того чтобы определить, какая из циклических систем образуется преимущественно при замыкании цикла, необходимо учитывать размер образующегося цикла и характер переходного состояния, приводящего к нему. Свободная энергия активации ![]() процесса состоит из энтальпийной (
процесса состоит из энтальпийной (![]() ) и энтропийной (
) и энтропийной (![]() ) компонент (Т – абсолютная температура):
) компонент (Т – абсолютная температура):
![]()
Энтропия активации для внутримолекулярного процесса связана с вероятностью подхода двух реакционных центров одной молекулы друг к другу. Эта вероятность уменьшается (![]() приобретает большое отрицательное значение) при увеличении длины цепи. Энтальпия активации отражает напряженность переходного состояния, приводящего к образованию цикла. Значение энтальпии активации наименьшее при образовании пяти- и шестичленных циклов и несколько увеличивается при образовании более напряженных трех- и четырехчленных циклических систем. Значение
приобретает большое отрицательное значение) при увеличении длины цепи. Энтальпия активации отражает напряженность переходного состояния, приводящего к образованию цикла. Значение энтальпии активации наименьшее при образовании пяти- и шестичленных циклов и несколько увеличивается при образовании более напряженных трех- и четырехчленных циклических систем. Значение ![]() велико при образовании циклов среднего размера (от восьми- до одиннадцатичленных), что связано с пространственными взаимодействиями в кольце. Значение свободной энергии активации при образовании циклов среднего размера также велико, поэтому замыкание таких циклов затруднено.
велико при образовании циклов среднего размера (от восьми- до одиннадцатичленных), что связано с пространственными взаимодействиями в кольце. Значение свободной энергии активации при образовании циклов среднего размера также велико, поэтому замыкание таких циклов затруднено.
Другой важный фактор при определении возможности протекания процесса замыкания цикла связан с геометрией подхода нуклеофильного центра к электрофильному в переходном состоянии. Хорошо известно, например, что при бимолекулярном нуклеофильном замещении при насыщенном атоме углерода реализуется переходное состояние, в котором нуклеофил приближается со стороны, противоположной уходящей группе. Атака нуклеофилом карбонильной группы предпочтительна сверху или снизу плоскости связи С=О под углом, близким к тетраэдрическому. Преимущественные направления атаки нуклеофилами различных электрофильных центров продемонстрированы на рис. 4. Вероятно, небольшие отклонения от подходящей геометрии допустимы, однако напряжение в некоторых типах процессов, приведенных на рис. 6, затрудняют образование циклов с пятью и меньшим количеством атомов. Например, можно предположить, что процесс эндо-триг «невыгоден» для образования циклов с числом атомов, меньшим шести. Действительно, такие реакции идут с большим трудом. Процесс эндо-диг с этой точки зрения еще более «невыгоден», но приводит к замыканию пятичленных циклов. Это возможно из-за того, что π-связи функциональной группы с sp-гибридизованным атомом углерода расположены в той же плоскости, что и нуклеофил, тогда как в эндотриг-процессе подход нуклеофила осуществляется сверху или снизу плоскости молекулы.
Замещение при насыщенном атоме углерода.
Внутримолекулярный вариант реакции SN2-типа широко используется для замыкания насыщенных гетероциклов. Наиболее легко этот процесс протекает при образовании пяти- и шестичленных циклов, поскольку при таком размере кольца наблюдается наилучший баланс между энтальпийной и энтропийной компонентами – кольца не напряжены и переходные состояния доступны. Циклизация бромоалкиламинов Br(CH2)n-1NH2 с образованием пяти- и шестичленных циклов особенно благоприятна. Азиридины (n = 3) образуются в результате этого процесса также достаточно легко, несмотря на существующее напряжение в трехчленном цикле. Малая степень упорядочения, требуемая для переходного состояния, облегчает протекание реакции. [Циклизация бромоацетат-анионов, приводящая к α-лактону, идет существенно медленнее, возможно, из-за возникновения дополнительного напряжения, связанного с включением sр2-гибридного атома углерода в цикл.]
Реакции этого типа сопровождаются инверсией при атоме углерода. На рис. 7 приведен пример стереоселективного образования цис- и транс-2,3-дифенилазиридинов из трео- и эритро-хлороаминов.
В тех случаях, когда скорость замыкания цикла мала, вероятно протекание межмолекулярных процессов и выход циклического продукта снижается. Например, взаимодействие 3-хлоропропанола с гидроксидом натрия в водном метаноле приводит к ациклическим продуктам сольволиза с выходом 78% и к оксетану лишь с выходом 14% (рис. 8).
Эффективность замыкания трех- и четырехчленных циклов увеличивается, если атом углерода, несущий нуклеофильную группу, максимально замещен. Это проиллюстрировано на примере образования оксиранов из этиленхлорогидринов (табл. 1). Увеличение скорости при введении заместителей связано с тем, что при образовании малого цикла величины валентных углов отклоняются от тетраэдрического, что уменьшает стерические затруднения.
Таблица 1. Относительные скорости реакций замыкания цикла для этиленхлорогидринов (водн. NaOH, 18 0C)
| Субстрат | Относительная скорость |
 |
1 |
 |
325 |
 |
39000 |
Cинтетическая значимость процессов замыкания цикла существенно возрастает при создании методов, позволяющих генерировать insitu подходящие предшественники циклических соединений. Для получения оксиранов из карбонильных соединений обычно используют два основных метода: реакцию Дарзана – взаимодействие карбонильных соединений с α-галогенокетонами и эфирами α-галогенокарбоновых кислот или синтезы с использованием илидов серы. Внутримолекулярное нуклеофильное замещение в образующихся интермедиатах приводит к замыканию оксиранового цикла.
В процессах замыкания цикла, основанных на внутримолекулярных SN-реакциях, используют в качестве нуклеофилов не только амино- и гидроксигруппы. В табл. 2 приведены примеры реакций, в которых еноляты кетонов и амиды в присутствии оснований выступают в качестве нуклеофилов.
Таблица 2. Примеры реакций замыкания цикла с использованием внутримолекулярного нуклеофильного замещения при насыщенном атоме углерода (все приведенные процессы замыкания цикла относятся к экзо-тет-типу).
| Реагенты | Интермедиат | Продукт реакции |
 |
 |
 |
 |
 |
 |
 |
 |
 |
Внутримолекулярное нуклеофильное присоединение к карбонильной группе.
Внутримолекулярное нуклеофильное присоединение к карбонильной группе широко используется для синтеза гетероциклических соединений. Нуклеофильная атака по карбонильной группе эфиров и хлороангидридов карбоновых кислот, а также аналогичных соединений сопровождается элиминированием уходящей группы, а карбонильная группа сохраняется в образующемся гетероцикле. Присоединение нуклеофила к карбонильной группе альдегидов и кетонов обычно влечет за собой дегидратацию образующегося циклического интермедиата, особенно в случаях, приводящих к гетероароматическим соединениям. При использовании слабых нуклеофилов циклизацию проводят при кислом катализе, в этом случае нуклеофил атакует активированную протонированием карбонильную функцию.
Различают три типа внутримолекулярного замыкания цикла с участием карбонильной групп альдегидов и кетонов: замыкание цикла по альдольному типу включает атаку нуклеофильным атомом углерода и приводит к гетероароматическому соединению; замыкание цикла происходит при нуклеофильной атаке гетероатомом; нуклеофильная атака орто-углеродного атома производных бензола приводит к бензоконденсированным гетероциклам.
Метод синтеза 2-замещенных индолов (табл. 3) основан на замыкании цикла при нуклеофильной атаке атомом азота аминогруппы карбонильной функции. Аминосоединение обычно не выделяют, а генерируют insitu при восстановлении. Также легко замыкание цикла идет при атаке гидроксильной и тиольной группами. Например, при синтезе изоксазолов взаимодействие β-дикарбонильного соединения с гидроксиламином приводит к монооксиму, который можно обнаружить в реакционной смеси. Дальнейшее замыкание цикла в монооксиме протекает довольно быстро и сопровождается элиминированием молекулы воды.
Таблица 3. Примеры циклизаций, включающих нуклеофильную атаку по карбонильной группе.
| Реагенты | Интермедиат | Продукт реакции |
а) Циклизация альдольного типа
|
 |
 |
б) Циклизация по нуклеофильным гетероатомам
|
 |
 |
в) Циклизация по орто-положению кольца
|
 |
 |



В большинстве методов синтеза бензоконденсированных гетероциклических соединений в качестве исходных соединений используют монозамещенные производные бензола. Свободное орто-положение бензольного кольца во многих случаях способно нуклеофильно атаковать электрофильный атом углерода карбонильной группы, расположенной в боковой цепи. Реакции такого типа обычно требуют кислотного катализа (протонные кислоты или кислоты Льюиса) для активации карбонильной группы. При синтезе хинолинов по Комба подходящий интермедиат для замыкания цикла получают insitu из анилина и дикетона. В противоположность этому, при синтезе бензофуранов обычно используют предварительно выделенный арилоксикетон. После замыкания цикла происходит быстрая дегидратация, приводящая к гетероароматическим соединениям.
Внутримолекулярное нуклеофильное присоединение к другим двойным связям.
Примеры замыкания цикла за счет нуклеофильного присоединения к двойным связям, отличным от С=О, приведены в табл. 4. В качестве электрофилов могут выступать активированные связи C=Sи C=N (пример 1). Аналогично может проявлять себя активированная связь С=С, в этом случае наблюдается внутримолекулярное сопряженное присоединение. Следует отметить, что в примере 2 реализуется кинетически предпочтительный экзо-триг-процесс, приводящий к замыканию четырехчленного цикла, а не возможный также эндо-триг-процесс образования пятичленного цикла.
Большинство методов синтеза гетероциклических соединений основано на циклизациях с участием электрофильного углеродного центра. Однако известно несколько аналогичных процессов с участием электрофильного атома азота. Пример 3 демонстрирует один из таких процессов, в которых в качестве электрофила выступает нитрогруппа.
Таблица 4. Другие примеры экзотриг-реакций.
| Реагенты | Интермедиат | Продукт реакции |
 |
 |
 |
 |
 |
 |
 |
 |
 |
К экзо-триг-процессам относятся также такие реакции, в которых замыкание цикла происходит в результате внутримолекулярного присоединения к неактивированной связи С=С. Большинство реакций подобного типа инициируется атакой по двойной связи внешним электрофилом. Образующийся при этом катионный интермедиат захватывает внутренний нуклеофил. Пример такой реакции приведен на рис. 9. В данном случае реакция инициируется бромом. Для инициирования подобных реакций используются также другие электрофилы: соли ртути (II), никеля (II) и других металлов, а также электрофильные органические производные селена, такие, как фенилселенилхлорид.
При синтезе пяти- и шестичленных гетероциклов, конденсированных с бензольным кольцом, используются циклизации, катализируемые палладием. Большинство процессов этого можно рассматривать как внутримолекулярный вариант реакции Чека, например синтез 3-метилиндола. Палладий, генерируемый insitu, катализирует такие процессы. Внедрение Pd(0) по связи углерод – галоген приводит к образованию палладийорганического интермедиата, который впоследствии присоединяется по двойной связи.
Циклизации с участием тригонального центра относятся главным образом к экзо-процессам. Однако существует важная группа эндо-процессов, приводящих к образованию пяти- и шестичленных азотсодержащих гетероциклов. Они основаны на генерировании и взаимодействии с внутренним нуклеофилом иминиевых солей. Простые иминиевые соли эффективно реагируют только с сильными нуклеофилами. N-ацилиминиевые соли – более сильные электрофилы, их можно получить несколькими различными способами. Взаимодействие N-ацилиминиевого катиона, полученного при восстановлении имида и последующей дегидратации, с внутренним нуклеофилом (связь С=С) приводит к замыканию новой циклической системы.
Электролитические реакции.
Все рассмотренные выше примеры замыкания цикла представляют собой внутримолекулярные версии хорошо известных реакций образования σ-связей. Электроциклические реакции существенно отличаются от этих примеров прежде всего тем, что не имеют межмолекулярных аналогов. Ациклические реагенты, используемые при электроциклическом замыкании цикла, должны представлять собой полностью сопряженные π-электронные системы. В электроциклических реакциях образование σ-связи происходит в результате преобразования π-системы. Нормальное течение процесса достигается при нагревании или облучении без участия дополнительных реагентов. Электроциклические процессы равновесны, причем равновесие обычно смещено в сторону ациклических изомеров, поэтому такие реакции более применимы для раскрытия, а не для замыкания цикла.
![]() Четыре типа электроциклических реакций, применяемых при синтезе гетероциклических соединений, схематически показаны на рис. 10. Примеры (а) и (б) иллюстрируют превращения сопряженных систем, содержащих четыре π-электрона. Замыкание цикла происходит либо в 1,3-диполе (а), либо в гетеродиене (б). Реакции (в) и (г) аналогичны (а) и (б), но цикл замыкается в шестиэлектронной π-системе. Таким образом, сопряженные π-электронные ациклические молекулы могут быть предшественниками насыщенных или частично насыщенных гетероциклов, содержащих от трех до шести атомов в цикле. Возможны также электроциклические процессы в сопряженных π-системах с числом электронов больше шести, хотя встречаются они гораздо реже.
Четыре типа электроциклических реакций, применяемых при синтезе гетероциклических соединений, схематически показаны на рис. 10. Примеры (а) и (б) иллюстрируют превращения сопряженных систем, содержащих четыре π-электрона. Замыкание цикла происходит либо в 1,3-диполе (а), либо в гетеродиене (б). Реакции (в) и (г) аналогичны (а) и (б), но цикл замыкается в шестиэлектронной π-системе. Таким образом, сопряженные π-электронные ациклические молекулы могут быть предшественниками насыщенных или частично насыщенных гетероциклов, содержащих от трех до шести атомов в цикле. Возможны также электроциклические процессы в сопряженных π-системах с числом электронов больше шести, хотя встречаются они гораздо реже.
Объяснение стереохимии реакций электроциклического замыкания и раскрытия циклов – первое достижение теории сохранения орбитальной симметрии, разработанной Вудвардом и Гофманом. Различают два типа электроциклизаций. В первом вращение р-орбиталей π-электронной системы осуществляется в одном направлении и приводит к образованию новой σ-связи, во втором – вращение происходит в разных направлениях. Первый процесс называют конротаторным (рис. 11, а), второй – дисротаторным (рис. 11, б). Правила Вудварда – Гофмана позволяют определить, какой из типов замыкания цикла предпочтителен, и тем самым предсказывают стереохимию образующегося циклического соединения. Правила определяются числом π-электронов, образующих сопряженную ациклическую систему, и тем, протекает ли процесс в основном состоянии (термическая реакция) или в первом возбужденном состоянии (фотохимическая реакция) полиена. Иллюстрация правил Вудварда – Гофмана приведена на рис. 11.
Любую электроциклическую реакцию можно осуществить как при нагревании (термически), так и при облучении (фотохимически), однако стереохимический результат в этих двух случаях будет различным. Синтетическая значимость электроциклических реакций существенно зависит от положения равновесия, поскольку это определяет, может ли циклический изомер быть выделен с удовлетворительным выходом из реакционной смеси. Положение этого равновесия может быть различным для термических и фотохимических процессов. Стереохимические различия между конротаторными и дисротаторными процессами исчезают в том случае, когда терминальное положение ациклической π-системы занимает гетероатом. Так, для большинства электроциклических процессов, приводящих к образованию гетероциклических соединений, правила Вудварда – Гофмана не имеют смысла.
Шестиэлектронные электроциклические процессы типа (в) (рис. 10) – 1,5-биполярное замыкание цикла – более распространены. В этом случае также наблюдаются равновесные процессы замыкания и раскрытия цикла. Нестабильные 1,5-диполи обычно получают insitu и при нагревании превращают в пятичленные гетероциклы. Циклический изомер может быть выведен из равновесия при таутомерном превращении в более стабильное (часто ароматическое) соединение. Примеры 1,5-диполярного замыкания цикла приведены в табл. 5. Пример 1 демонстрирует замыкание цикла в нестабильном 1,5-диполе. В примерах 2 и 3 первоначально образующийся продукт циклизации тау-томеризуется в ароматическое соединение, что смещает равновесие в сторону циклического изомера. Реакции 4 и 5 сразу приводят к образованию ароматических гетероциклов.
Таблица 5. Примеры 1,5-диполярной циклизации.
| Исходное соединение | 1,5-диполярный интермедиат | Продукт циклизации | Конечный продукт (если он отличается) |
| 1. |
 |
 |
_____ |
| 2. Винилдиазометан не может быть выделен, но циклизуется при t>250C. |  |
 |
 |
3.  |
 |
 |
 |
4.  |
 |
 |
_____ |
5.  |
 |
 |
_____ |
Электроциклические процессы в гетеротриенах [реакция (г), рис. 11)] могут быть применены при синтезе некоторых шестичленных гетероциклов, особенно в тех случаях, когда продукт циклизации способен к ароматизации. Для шестиэлектронных электроциклических процессов, так же как и в других случаях, возможна обратная реакция раскрытия цикла. В некоторых случаях этот обратный процесс более важен с синтетической точки зрения.
В табл. 6 приведены некоторые примеры синтеза шестичленных гетероциклов с использованием электроциклических процессов Синтез изохинолинов (реакция 4) основан на двух электроциклических реакциях: образовании орто-ксилиленового интермедиата при раскрытии бензоциклобутана и 6π-электронном электроциклическом процессе. Замыкание цикла на второй стадии идет очень легко, так как при этом восстанавливается ароматичность бензольного кольца.
В некоторых случаях электроциклические процессы в гетеротриенах приводят к образованию пятичленных гетероциклов. Вероятность протекания таких процессов увеличивается при наличии электроотрицательного гетероатома в терминальном положении гетеродиена.
Таблица 6. Образование шестичленных гетероциклов электроциклизацией.
| Исходное соединение | Интермедиат | Продукт реакции |
1.  , нагревание , нагревание |
_______ |  |
2.  , нагревание , нагревание |
 |
 |
| 3. |
 |
 |
4. 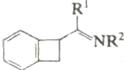 , нагревание , нагревание |
 |
 |
5.  |
 |
 |
Акридин.
Общие сведения. Акридин является дибензопиридином и относится к хинолину так же, как антрацен к нафталину. Его можно рассматривать как антрацен, в котором одна центральная группа =СН- замещена азотом.
Нумерация атомов производится следующим образом:


III
В ряде стран, в том числе в России, принята нумерация I, в других странах (Англия, США) – нумерация II.
Акридин открыт в 1870г. в неочищенной антраценовой фракции каменно-угольного дегтя. Он плавится при 107οС, кипит при 345-346οС, легко возгоняется. Обладает характерным запахом, вызывает раздражение дыхательных путей, раздражает кожу, откуда и произошло название (acer – едкий). Акридин светится при трении (триболюминисценция). Разбавленные растворы имеют синюю флуоресценцию. Соли акридина в разбавленных растворах обладают зеленой флуоресценцией. При дальнейшем разбавлении, вызывающем гидролиз, флуоресценция переходит в синюю, характерную для свободного акридина. Акридиновое ядро составляет основу некоторых алкалоидов. Многие синтетические производные акридина являются ценными лекарственными препаратами и красителями.
Способыполучения. Несмотрянато,чтоакридин содержится в каменноугольной смоле, его производныеисамакридинполучаютсинтетически из производных бензола.
1. Наиболееобщимспособом получения акридина является циклизация о-анилинобензойной кислоты. Замыкание цикла может быть достигнуто с помощью серной кислоты, и в этом случае образуется акридон. Он может быть восстановлен амальгамой натрия в дигидроакридин (акридан), который затем окисляют хлоридом железа в акридин (рис. 12).
Однако чаще всего циклизацию проводят с помощью РОСl3 получая 9-хлоракридин (рис. 13).
Хлор, находящийсявмезо-положении, отличаетсябольшойреакционнойспособностью илегкоможетбытьзамещеналкокси-, аминогруппойиливодородом. Используязамещенныео-анилинобензойныекислоты, можнополучитьразличныепроизводныеакридина.
2. Конденсациям-фенилендиаминасмуравьинойкислотойилиформальдегидомпозволяетполучать 3- и 6-диаминоакридины:
Химическиесвойства. Акридинявляетсяслабымоснованием, образуетсоли, такие, как хлорид, нитрат, пикрат и т.п., окрашенные четвертичные аммониевые соли (соединения акридиния) и N-оксид (при обработке гидропероксидом бензоила).
Акридинкипитпривысокойтемпературебезразложенияинеизменяетсяприсплавленииседкимкали. Окислениеперманганатомчастичноразрушаетмолекулусобразованием акридиновойкислоты(2,3-хинолиндикарбоноваякислота) (рис. 14, I), однакодихроматвуксусной кислотенеразрушаетциклическуюструктуруакридина, нопревращаетеговакридон (рис. 14, II) и 10,10'-диакридоннл (рис. 14, III).
Междуакридиномиантраценомимеетсясходство. Оновыражаетсявспособностиакридинаквосстановлениюводородомвмоментвыделения (придействиинатриявводныхи спиртовыхрастворахилиприкаталитическомгидрированиив 9,10-дигидроакридин. Присоединениенатриясобразованием 9,10-динатриевогопроизводного, котороесоспиртомдает акридан, напоминаетреакциюнатриясантраценом.
Некоторыереакцииакридинанапоминаютреакциипиридинаиегопроизводных. Литий-алкилыприсоединяютсякакридинувположения 9, 10, т.е. поконцамсопряженнойсистемы. Последующийгидролиздаетпроизводное 9,10-дигидроакридина.
Мезо-метилакридин, подобно 4-пиколину, конденсируетсясальдегидами.
Производные акридина. Из производных акридина важное применение имел так называемый акрихин – 6-хлор-2-метокси-9-(1-метил-4-диэтиламино) бутиламиноакридин дихлоргидрат – желтое кристаллическое вещество с т. пл. 248—250 °С.
Акрихин широко использовался для предупреждения и лечения малярии. Он в известной степени является заменителем хинина.
Риванол — соль молочной кислоты акридинового производного следующего строения:
Эффективный антисептик. Применяется также для лечения амебной
дизентерии.
3. ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Таким образом, в предыдущих разделах рассмотрен материал, включающий различные методы получения гетероциклических соединений реакциями замыкания цикла, свойства, общую информацию, применение и получение акридина; свойства, методы синтеза и применение акридона.
Акридон был получен кипячением смеси фенилантраниловой кислоты с серной. Выход продукта составил 1,78г или 74% от теоретического выхода. Температура плавления полученного соединения составила 3440С, что полностью совпадает со справочными данными. Подобное совпадение свидетельствует о чистоте полученного продукта.
На основании собранной теоретической информации можно выдвинуть предположительный механизм данной реакции:
4. ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты и оборудование.
Уравнение реакции имеет вид:

Реактивы: N-фенилантраниловая кислота (С13Н11О2N, M=213 г/моль, Тпл=179-1810С) – 3г, серная кислота (Н2SО4, M=98.08 г/моль, Тпл=-13.60С, Ткип=301,30С, плотность ρ=1,85, концентрация – 0,96%) – 7мл. Карбонат натрия – 2,12г (0,02моль). Вода.
Теоретический выход вещества по взятому в недостатке реактиву, участвующему в реакции (N-фенилантраниловая кислота) составляет 2,4г.
Схема прибора:
Методика эксперимента. В круглодонной колбе емкостью 50мл приготовлен раствор 3г N-фенилантраниловой кислоты в 7мл серной кислоты (98%) и раствор нагревался на кипящей водяной бане в течение 4-х часов, после чего его вылили в 100мл кипящей воды. Раствор приливали по стенке сосуда. В течение 5мин кипятили смесь, затем желтый осадок отфильтровали, сохранив фильтрат. Влажный осадок прокипятили в течение 5мин с раствором 2,12г (0,02 моль) соды в 30мл воды, после чего его отсосали с помощью водоструйного насоса, колбы Бунзена и воронки Бюхнера и хорошо промыли водой. Затем влажный осадок был высушен на воздухе. Выход вещества составил 1,78г или 74% от теоретического. Измеренная точка плавления вещества составила примерно 3440.
5. ВЫВОДЫ
Таким образом, в данной работе рассмотрен акридон, его свойства, способы получения и применение. Приведены различные механизмы получения гетероциклических соединений реакциями конденсации. Предложен возможный механизм реакции синтеза акридона. Кипячением N-фенилантраниловой кислоты с концентрированной серной получен акридон с выходом 1,78г или 74% масс. от теоретического. Измеренная точка плавления показала сходимость полученных данных с литературными, а следовательно, очень низкую долю примесей в полученном акридоне и чистоту продукта синтеза.
6. БИБЛИОГРАФИЧЕСКИЙ СПИСОК
1. Воробьев А.А. Принципы классификации и стратегия применения иммуномодуляторов в медицине. ЖМЭИ, 2002, N4, c.93-98.
2. ColonnaM., Krug A., Cella M. Interferon-producing cells: on the front line in immune responses against pathogens. Curr.Opin.Immunol. 2002, 14, 373-379.
3. Джилкрист Т. Химия гетероциклических соединений. М.: Мир, 1996, с.78-109, 203-205.
4. Петров А.А., Бальян Х.В., Трощенко А.Т. Химия органических соединений, учебник для ВУЗов. С.-Пб.: Иван Федоров, 2002, с.521-524.
5. Лернер И.М., Гонор А.А., Славачевская Н.М., Берлин А.И. Указатель препаративных синтезов органических соединений. 2-ое изд., перераб. и доп. Л.: Химия, 1982.
6. Свойства органических соединений: справочник /под ред. А.А. Потехина, Л.: Химия,1984.
7. Шабаров Ю.С. Органическая химия, учебник для ВУЗов: в 2 кн. М.: Химия, 1996.
8. Земцова М.Н. Органическая химия. Указания к курсовой работе. Самар. гос. техн. ун-т. Самара, 2005, с.23.