| Скачать .docx |
Реферат: Влияние модифицированной полиметакриловой кислоты, ковалентно связанной с порфирином, на его кислотно-основные свойства
Среди разнообразных моделей первичных процессов фотосинтеза в последнее время важные результаты по стабилизации разделенных зарядов получены с водорастворимыми порфиринами, содержащими ионогенные группы, в частности положительно заряженными порфиринами Zn [1—3]. Имеются данные по использованию полиэлектролитов для стабилизации разделенных зарядов за счет их взаимодействия с электростатическим полем в макроионах. Однако эти вопросы в полимерных системах недостаточно разработаны [4—6]. Можно ожидать, что иммобилизация порфиринов на полимерах должна приводить к изменению ряда свойств этих макроциклических молекул. В частности, в ионогенных водорастворимых полимерах, содержащих гидрофобные области, возможно изменение кислотно-основных свойств порфирина, как в случае изменения па два — три порядка основности алифатических аминогрупп в водном растворе при иммобилизации их в гидрофобных областях полимерных молекул [7].
Цель настоящей работы состояла в том, чтобы выяснить, в какой мере различное полимерное микроокружение оказывает влияние на реакцию протонирования Ш-тетра (аминофенил) порфина (Н2 ТАФП), ковалентно связанного с водорастворимыми полимерами. Такая формулировка задачи становится возможной, поскольку связанный с водорастворимым полимером порфирин приобретает растворимость в водной среде, что позволяет избежать модификации его молекулы ионогенными группами.
Н2 ТЛФП марки х.ч. использовали без дополнительной очистки. Химически связанный с полиметакриловой кислотой Н2 ТАФП (И2 ТАФП — ПМАК) получали реакцией взаимодействия порфирина с ПМАК, содержащей ~1% хлорангидридных групп [8]. Введение цетильных групп в Н2 ТАФП — ПМАК осуществляли амидированием карбоксильных групп цетиламином в присутствии дициклогексилкарбодина ч.д.а. [9]. Для повышения гидрофобности порфирина, а также предотвращения сшивании полимера проводили реакцию свободных аминогрупп Н2 ТАФП, иммобилизованного па ПМАК, с бромистым гексилом. Очистку полимера осуществляли путем диализа, а выделение — последующей лиофильной сушкой. Содержание порфирина в обоих полимерах определяли в 2-10~2 м. растворе триэтиламина, рН~11, спектрофотометрически, по интегральной интенсивности полосы Соре. В Н2 ТАФП — ПМАК мольное (в расчете на мономерное звено) отношение Н2 ТАФП : МАК= = 1:1800, в Н2 ТАФП — ПМАК-цетил оно составляет 1:150. Количество цетильных групп в макромолекулах Н2 ТАФП — ПМАК-цетил определяли по соотношению интенсивностей колебаний амидной и карбоксильной групп в ПК-спектрах при 1570 и 1700 см-1 . Количество цетильных групп составило 46% от мономерных звеньев ПМАК. По данным вискозометрии, молекулярная масса Н2 ТАФП — ПМАК-цетил, измеренная в 0,002 н. НС1, составляла 9-10*.
Коэффициент экстинкции димерного Н2 ТАФП был получен из анализа спектров поглощения ряда растворов с различным соотношением димер : мономер, в которых исследовали взаимосвязь коэффициента экстинкции и полуширины полосы Соре. За коэффициент экстинкции димерного порфирина в расчете на мономер было принято значение, при котором с увеличением количества димеров в растворе относительное уширение полосы Соре не сопровождалось изменением коэффициента экстинкции.
Спектры поглощения снимали на спектрофотометре «Specord М-40» (ГДР). Измерение рН проводили с помощью универсального иономера ЭВ-74.
Реакции протонирования и депротонирования Н2 ТАФП в растворах происходят путем последовательного присоединения или удаления протонов от центральных атомов азота порфиринового кольца. Равновесие этих реакций изучали методом спектрофотометрического титрования на двух образцах

При титровании водного раствора Н2 ТАФП — ПМАК из кислой области раствором КОН в спектре порфирина наблюдается снижение интенсивности полос поглощения с λмакс =445 и 654 нм. Одновременно возрастает интенсивность полосы с λмакс =420 нм (рис. 1). Спектральные изменения сопровождаются появлением четырех изобестических точек при длинах волн 426, 464, 579 и 698 нм, свидетельствующих о том, что с увеличением рН в системе происходит изменение концентраций двух типов поглощающих центров, которыми являются протонированная и непротонированная формы Н,ТАФП. Спектр депротонированного порфирина по характеристикам соответствует спектру мономера. Используя эффективный коэффициент экстинкции протонированнной формы порфирина (еэф =0,44-105 л/моль-см) при Х=654 нм, который определяли в области рН~3, где порфирин полностью протонирован, поскольку понижение рН не приводило к изменению спектра, получили зависимость концентрации протонированной формы порфирина от рН (рис. 2). Наличие двух S-образных переходов на кривой позволяет установить две константы равновесия, которые определяли в точках, когда степень диссоциации а титрующейся системы равна 0,5, используя при этом уравнение Гендерсона — Хассельбаха
pK = pH + lg
Найденные значения констант равны рК, 5,6 и рК2 7,9. Как видно из рис. 2, при низких рН оттитровывается только 8% молекул порфирина. Процесс депротонирования этих молекул происходит в области рН 4,5— 5,7, т. е. там, где оттитровывается от 10 до 22% ПМАК (рис. 2, кривая2). Это область рН, когда в ПМАК осуществляется конформационный переход. Макромолекулы из стабилизированной водородными связями и гидрофобными взаимодействиями цепей вторичной конформацин переходят в конформацию асимметричных рыхлых отрицательно заряженных клубков. Остальная часть порфирина титруется, уже находясь в поле отрицательно заряженного полианиона. В результате увеличивается рКа диссоциации протонированного порфирина более чем на две единицы рН, до 7,9, поскольку, для того чтобы вывести протоны из клубка, необходимо преодолеть электростатическое поле заряженной макромолекулы.

Рис. 1. Спектральные изменения Н2 ТАФП, связанного с ПМАК, при различных рН водного раствора. Стрелками показано направление изменения рН от 7,2 до 10,84

Рис. 2. Зависимость количества ионизованных молекул порфина с и степени ионизации а ПМАК (2) от рН раствора
Весьма существенно, что наблюдаемый эффект роста рКа за счет электростатического взаимодействия в полигоне значительно больше, чем у отрицательно заряженных порфиринов (таблица). Наиболее низкое значение рК соответствует тетрасульфопроизводному порфирина, оно близко по величине к рК порфирина в ПМАК, несущей от 10 до 22% заряженных карбоксильных групп. Этот результат свидетельствует о том, что в полимере карбоксилат-анионы сближены сильнее с протонами порфирина, чем анионы в молекуле Н2 ТФП(803 )4 4_ , в которой заряды расположены по периферии молекулы на расстоянии 7—9 А от ее центра, где локализован положительный заряд. Еще большая величина рК Н2 ТКФП~, по-видимому, связана не с влиянием карбоксилат-аниона, а с тем, что порфирины были помещены в мицеллы додецилсульфата, величина суммарного отрицательного заряда которых весьма значительна. Однако и в этом случае эффект повышения рК не столь высок, как в макромолекулах ПМАК, заряженных на 80%, где имеется хорошая возможность для локального сближения молекул порфирина с карбоксильными анионами.
Спектр поглощения порфирина, связанного с полимером, содержащим 46% цетильных групп, указывает на присутствие в системе как мономерного, так и димерного порфирина. При подкислении водного раствораН2 ТАФП — ПМАК-цетил раствором НС1 наблюдается уменьшение интенсивности полосы Соре при А,макс =427 нм и одновременно увеличение интенсивности полосы поглощения с Ама1(С =715 нм (рис. 3). Наличие изобестичных точек при А,=442, 515 и 581 нм и характер изменения поглощения свидетельствуют о том, что по мере протонирования порфирина происходит изменение концентраций двух поглощающих центров, т. е. соотношение между мономерами и димерами при протонировании порфирина не нарушается. С использованием данных по поглощению при 715 нм и общей концентрации частиц порфирина (мономерных и димерных) был определен эффективный коэффициент экстинкции протонированного порфирина (еэф =0,45 1 05 л/моль*см). Зависимость концентрации протонированного порфирина от рН в макромолекуле ПМАК-цетил представлена на рис. 4, а.
Определение числа протонов, участвующих в кислотно-основном равновесии, позволило бы установить характер протонирования порфирина. Для этого используем выражение для суммарной константы диссоциации заряженного порфирина
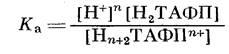
Логарифмируя это выражение, получим
![]()
Если область кривой титрования, соответствующую переходу непротонированного порфирина в протонированный, построить в координатах уравнения (2), то при условии выполнения линейной зависимости можно определить суммарное число протонов п, участвующих в равновесии. Значение п, определенное из представленной на рис. 4, б прямолинейной зависимости, равно двум. Следовательно, протонирование порфирина в ПМАК-цетил осуществляется по двум атомам азота. В щелочных средах (рН~11), когда макромолекулы ПМАК-цетил заряжены, их размеры по данным квазиупругого рассеяния света составляют 700 А без ионной силы и 600 А в 0,05 м. растворе КС1. Поскольку в водной среде цетильные группы, среднее количество которых на макромолекулу составляет ~250, образуют гидрофобные области, состоящие из нескольких десятков цетильных групп, то в среднем в макромолекуле присутствуют две — четыре гидрофобные области. Можно полагать, что мономерные и димерные молекулы Н2 ТАФП локализованы именно в этих областях вследствие высокой гидрофобности самого порфирина, и тем более порфирина, алкилированного по свободным аминогруппам бромистым гексилом. Константа кислотно-основного равновесия Н2 ТАФП, расположенного в гидрофобной области, должна зависеть от двух факторов: степени гидрофобности микроокружения и наличия заряда на самом порфирине или в ближайшем окружении. Как показано в таблице, порфирины, несущие четыре положительных заряда, имеют рКа в водном растворе более низкие по сравнению с рКа незаряженного Н2 ТАФП в бензоле, что свидетельствует о значительном электростатическом влиянии положительного заряда на рКа порфирина. Еще более сильное влияние заряда на рКа порфирина найдено у пиридинсодержащих молекул, где положительный заряд делокализован по системе сопряжения макроциклических молекул.


Рис. 3. Спектральные изменения Н2 ТАФП, связанного с макромолекулой ПМАК-цетил при различных рН водного раствора. Стрелками показано направление изменения рН от 11,7 до 0,06
Рис. 4. Зависимости количества ионизованных молекул порфирина с от рН раствора (о) и lgJ от рН водного раствора Н2 ТАФП - ПМАК-цетил (б), а: 1 — водный раствор Н2 ТАФП - ПМАК-цетил; 2 - водный диметилформамидный (1 :1) раствор Н2 ТАФП -ПМАК-цетил; б: ИН2 ТАФП] : [НП+2 ТАФП"+]
Процесс протонирования Н2 ТАФП в полимере начинается в области рН 2,5 и заканчивается при рН 0,2 (рис. 4, а). Значение константы равновесия, соответствующее этой области рН (по уравнению (1)), равно0,83. Его можно связать как с гидрофобностыо микроокружения порфирина в полимере, так и присутствием в его молекуле одноименных зарядов, т. е. с обоими факторами, понижающими основность порфирина. Гидрофобность определяется детальными группами на полимере, а заряды на порфирине возникают при протонировании его трех алкилированных аминогрупп. Гидрофобные взаимодействия в Н2 ТАФП — ПМАК-цетил могут быть ослаблены добавлением в систему смешивающегося с водой органического растворителя. Следствием этого должно быть возрастание гидратации порфирина и соответственно увеличение рКа . На рис. 4, а приведена кривая титрования порфирина, связанного с ПМАК-цетил в смеси Н2 0:ДМФА-1 : 1. В этих условиях рКа Н2 ТАФП увеличивается до 4,65, т. е. принимает промежуточное значение между рассмотренными случаями.
Таким образом, следует заключить, что в полимерных системах может реализовываться микроокружение молекул порфиринов, изменяющее их рКа на 7 единиц. Варьируя микроокружение, можно в широком интервале изменять основность порфирина. Изменение кислотно-основных свойств порфирина в полимерном микроокружении проявляется существенно сильнее, чем у низкомолекулярных систем.
ЛИТЕРАТУРА
1. Richoux М.-С, Harriman А. II1. Chem. Soc. Faraday Trans. I. 1982. V. 78. P. 1873.
2. Harriman A., Porter G., Richoux М.-С. ЦJ. Chem. Soc. Faraday Trans. II. 1981. V. 77. P. 833.
3. Blondeel G., Harriman A., Porter G., Wilowska А.Ц1. Chem. Soc. Faraday Trans. II. 1984. V. 80. P. 867.
4. Utvos J. W., Casti Т. E., Calvin M. // Sci. Papers Inst. Phys. and Chem. Research. V. 78. 1984. № 4. P. 129.
5. Meyerstein D., Radani J., Matheson M. S., Meisel D.//J. Phys. Chem. 1978. V. 82. № 17. P. 1879.
6. Mango M., Dannhauser Т., Huang D., Spears K., Morrison L., Leach P. A. // Macromo-lecules. 1984. V. 17. P. 1898.
7. Пшежецкий В. С, Лукьянова А. П. // Биоорг. химия. 1976. Т. 2. № 1. С. 110.
8. Риш И. Г., Пономарев Г. В., Белъговский И. М., Аскаров К. А., Пшежецкий В. С. Ц IV Всесоюз. конф. по химии и применению порфиринов. Ереван, 1984. С. 59.
9. Физер Л., Физер М. Реагенты для органического синтеза. Т. 1. М.. 1970. С. 422.
10. Harriman A., Richoux М. С. // J. Photochem. 1984. V. 27. Р. 205.
11. Williams С. N., Hambright Р. // Inorgan. Chem. 1978. V. 17. № 9. P. 2687.
12. Shamin A., Hambright P. // Inorgan. Chem. 1980. V. 19. № 2. P. 564.