| Скачать .docx |
Реферат: Вычисление теплового эффекта реакций
7. Вычислить тепловой эффект реакции при стандартных условиях: Fe2 O3 (т) + 3 CO (г) = 2 Fe (т) + 3 CO2 (г) ,если теплота образования: Fe2 O3 (т) = – 821,3 кДж/моль;СО(г) = – 110,5 кДж/моль;
СО2 (г) = – 393,5 кДж/моль.
Fe2 O3 ( т ) + 3 CO ( г ) = 2 Fe ( т ) + 3 CO2 ( г ) ,
Решение.
Зная стандартные тепловые эффекты сгорания исходных веществ и продуктов реакции, рассчитываем тепловой эффект реакции при стандартных условиях:

16. Зависимость скорости химической реакции от температуры. Правило Вант-Гоффа. Температурный коэффициент реакции.
К реакциям приводят только столкновения между активными молекулами, средняя энергия которых превышает среднюю энергию участников реакции.
При сообщении молекулам некоторой энергии активации Е (избыточная энергия над средней) уменьшается потенциальная энергия взаимодействия атомов в молекулах, связи внутри молекул ослабевают, молекулы становятся реакционноспособными.
Энергия активации не обязательно подводится извне, она может быть сообщена некоторой части молекул путем перераспределения энергии при их столкновениях. По Больцману, среди N молекул находится следующее число активных молекул N обладающих повышенной энергией :
N N·e– E / RT (1)
где Е – энергия активации, показывающая тот необходимый избыток энергии, по сравнению со средним уровнем, которым должны обладать молекулы, чтобы реакция стала возможной; остальные обозначения общеизвестны.
При термической активации для двух температур T1 и T2 отношение констант скоростей будет:
 , (2)
, (2)
откуда , (3)
, (3)
что позволяет определять энергию активации по измерению скорости реакции при двух различных температурах Т1 и Т2 .
Повышение температуры на 100 увеличивает скорость реакции в 2 – 4 раза (приближенное правило Вант-Гоффа). Число, показывающее, во сколько раз увеличивается скорость реакции (следовательно, и константа скорости) при увеличении температуры на 100 называется температурным коэффициентом реакции:
 (4)
(4)
Или .(5)
.(5)
Это означает, например, что при увеличении температуры на 1000 для условно принятого увеличения средней скорости в 2 раза ( = 2) скорость реакции возрастает в 210 , т.е. приблизительно в 1000 раз, а при = 4 –в 410 , т.е. в 1000000 раз. Правило Вант-Гоффа применимо для реакций, протекающих при сравнительно невысоких температурах в узком их интервале. Резкое возрастание скорости реакции при повышении температуры объясняется тем, что число активных молекул при этом возрастает в геометрической прогрессии.
25. Уравнение изотермы химической реакции Вант-Гоффа.
В соответствии с законом действующих масс для произвольной реакции
а A + bB = cC + dD
уравнение скорости прямой реакции можно записать:
![]() ,
,
а для скорости обратной реакции: ![]() .
.
По мере протекания реакции слева направо концентрации веществ А и В будут уменьшаться и скорость прямой реакции будет падать. С другой стороны, по мере накопления продуктов реакции C и D скорость реакции справа налево будет расти. Наступает момент, когда скорости υ 1 и υ 2 становятся одинаковыми, концентрации всех веществ остаются неизменными, следовательно,
![]() ,
,
ОткудаKc
= k1
/ k2
= ![]() .
.
Постоянная величина Кс , равная отношению констант скоростей прямой и обратной реакций, количественно описывает состояние равновесия через равновесные концентрации исходных веществ и продуктов их взаимодействия (в степени их стехиометрических коэффициентов) и называется константой равновесия. Константа равновесия является постоянной только для данной температуры, т.е.
Кс = f (Т). Константу равновесия химической реакции принято выражать отношением, в числителе которого стоит произведение равновесных молярных концентраций продуктов реакции, а в знаменателе – произведение концентраций исходных веществ.
Если компоненты реакции представляют собой смесь идеальных газов, то константа равновесия (Кр ) выражается через парциальные давления компонентов:
Kp
= ![]() .
.
Для перехода от Кр к Кс воспользуемся уравнением состояния P · V = n·R·T. Поскольку
![]() , то P = C·R·T.
, то P = C·R·T.
Тогда ![]() .
.
Из уравнения следует, что Кр = Кс при условии, если реакция идет без изменения числа моль в газовой фазе, т.е. когда (с + d) = (a + b).
Если реакция протекает самопроизвольно при постоянных Р и Т или V и Т, то значенияG и F этой реакции можно получить из уравнений:
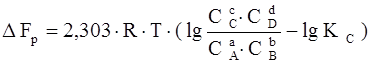 ,
,
где С А , С В , С С , СD – неравновесные концентрации исходных веществ и продуктов реакции.
 ,
,
где Р А , Р В , Р С , РD – парциальные давления исходных веществ и продуктов реакции.
Два последних уравнения называются уравнениями изотермы химической реакции Вант-Гоффа. Это соотношение позволяет рассчитать значения G и F реакции, определить ее направление при различных концентрациях исходных веществ.
Необходимо отметить, что как для газовых систем, так и для растворов, при участии в реакции твердых тел (т.е. для гетерогенных систем) концентрация твердой фазы не входит в выражение для константы равновесия, поскольку эта концентрация практически постоянна. Так, для реакции
2 СО (г) = СО 2 (г) + С (т)
константа равновесия записывается в виде
 .
.
Зависимость константы равновесия от температуры (для температуры Т2 относительно температуры Т1 ) выражается следующим уравнением Вант-Гоффа:
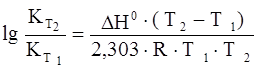 ,
,
где Н0 – тепловой эффект реакции.
Для эндотермической реакции (реакция идет с поглощением тепла) константа равновесия увеличивается с повышением температуры, система как бы сопротивляется нагреванию.
34. Осмос, осмотическое давление. Уравнение Вант-Гоффа и осмотический коэффициент.
Осмос – самопроизвольное движение молекул растворителя через полупроницаемую мембрану, разделяющую растворы разной концентрации, из раствора меньшей концентрации в раствор с более высокой концентрацией, что приводит к разбавлению последнего. В качестве полупроницаемой мембраны, через маленькие отверстия которой могут селективно проходить только небольшие по объему молекулы растворителя и задерживаются крупные или сольватированные молекулы или ионы, часто служит целлофановая пленка – для высокомолекулярных веществ, а для низкомолекулярных – пленка из ферроцианида меди. Процесс переноса растворителя (осмос) можно предотвратить, если на раствор с большей концентрацией оказать внешнее гидростатическое давление (в условиях равновесия это будет так называемое осмотическое давление, обозначаемое буквой ). Для расчета значения в растворах неэлектролитов используется эмпирическое уравнение Вант-Гоффа:
= CRT,
где С – моляльная концентрация вещества, моль/кг;
R – универсальная газовая постоянная, Дж/моль · К.
Величина осмотического давления пропорциональна числу молекул (в общем случае числу частиц) одного или нескольких веществ, растворенных в данном объеме раствора, и не зависит от их природы и природы растворителя. В растворах сильных или слабых электролитов общее число индивидуальных частиц увеличивается вследствие диссоциации молекул, поэтому в уравнение для расчета осмотического давления необходимо вводить соответствующий коэффициент пропорциональности, называемый изотоническим коэффициентом.
i · C · R · T,
где i – изотонический коэффициент, рассчитываемый как отношение суммы чисел ионов и непродиссоциировавших молекул электролита к начальному числу молекул этого вещества.
Так, если степень диссоциации электролита, т.е. отношение числа молекул, распавшихся на ионы, к общему числу молекул растворенного вещества, равна и молекула электролита распадается при этом на n ионов, то изотонический коэффициент рассчитывается следующим образом:
i = 1 + (n – 1) · ,(i > 1).
Для сильных электролитов можно принять = 1, тогда i = n, и коэффициент i (также больше 1) носит название осмотического коэффициента.
Явление осмоса имеет большое значение для растительных и животных организмов, поскольку оболочки их клеток по отношению к растворам многих веществ обладают свойствами полупроницаемой мембраны. В чистой воде клетка сильно набухает, в ряде случаев вплоть до разрыва оболочки, а в растворах с высокой концентрацией солей, наоборот, уменьшается в размерах и сморщивается из-за большой потери воды. Поэтому при консервировании пищевых продуктов к ним добавляется большое количество соли или сахара. Клетки микроорганизмов в таких условиях теряют значительное количество воды и гибнут.
Осмотическое давление обеспечивает движение воды в растениях за счет различия осмотических давлений между клеточным соком корней растений (5-20 бар) и почвенным раствором, дополнительно разбавляемом при поливе. Осмотическое давление обусловливает в растении подъем воды от корней до вершины. Таким образом, клетки листьев, теряя воду, осмотически всасывают ее из клеток стебля, а последние берут ее из клеток корня.
43. Рассчитать эквивалентную электропроводность 1 н раствора Na NO3 , если его удельное сопротивление равно 12,42 Ом · см.
Решение.
Эквивалентная электропроводность – величина электропроводности, отнесенная к одному моль – эквиваленту электролита:
 ,
,
где λ – эквивалентная электропроводность, Ом – 1 · см2 · моль– 1 ;
Сэк – молярная концентрация эквивалентов раствора электролита, моль/л.
Удельная электропроводность – величина, обратная удельному сопротивлению:
![]()
где cудельная электрическая проводимость, Ом – 1. см – 1 ;
ρ – удельное электрическое сопротивление, Ом · см.
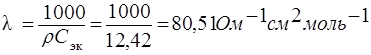
52. Отличительные признаки дисперсных систем от истинных растворов. Механизм проявления каждого отличительного признака.
Истинный раствор — это гомогенные смеси, состоящие из растворенных веществ и растворителя. В истинных растворах растворенные вещества находятся либо в молекулярно-дисперсном, либо в ионно-дисперсном состоянии.
Истинно растворимые частицы обуславливают, в частности, осмотическое давление, осмотические явления снижения температуры замерзания и повышения температуры кипения.
Высокодисперсные гетерогенные системы, в отличие от растворов, содержат чаще всего 2 фазы. Одна фаза представляет собой высокодисперсные мельчайшие частицы вещества или макромолекулы ВМС и называется дисперсной фазой. Другая фаза, в которой распределены агрегаты дисперсных частиц или макромолекул, называется дисперсионной средой. Условием образования таких дисперсных систем (коллоидного состояния вещества) является нерастворимость одной фазы в другой.
Дисперсная фаза, состоящая из множества мельчайших частиц, имеет очень большую удельную поверхность раздела с дисперсионной средой. Особые свойства поверхности раздела обусловливают специфические особенности дисперсных систем, что и является причиной выделения данной области знания в отдельную науку – коллоидную химию.
Основными отличительными особенностями дисперсных систем от истинных растворов являются:
а) способность к рассеиванию ими света;
б) медленная диффузия частиц дисперсной фазы в дисперсионной среде;
в) способность к диализу;
г) агрегативная неустойчивость дисперсной фазы, которая определяется выделением частиц из дисперсионной среды при добавлении к системе электролитов или под влиянием других внешних воздействий.
61. Рассчитать средний сдвиг частиц аэрозоля с радиусом частиц 10-7 м за время 10 с при температуре 273 К и вязкости воздуха 1,7·10-5 н·с/м2 . Как изменится средний сдвиг частиц, если радиус частиц аэрозоля увеличится до 10-6 м?
Средний сдвиг частиц аэрозоля
![]()
время, за которое происходит смещение частицы (продолжительность диффузии), с;
D коэффициент диффузии, м2 . с-1 .
Коэффициент диффузии для сферической частицы рассчитывается по уравнению Эйнштейна:
 ,
,
где NА – число Авогадро, 6 10 23 молекул/моль;
h – вязкость дисперсионной среды, Н с/м2 (Па с);
r – радиус частицы, м;
R – универсальная газовая постоянная, 8,314 Дж/моль · К;
T – абсолютная температура, К;
число 3,14.
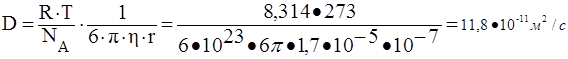
![]()
ответ на второй вопрос задания:

![]()
Таким образом, средний сдвиг частицы уменьшиться в 10 раз.
Ответ: ![]() , уменьшится в 10 раз.
, уменьшится в 10 раз.
80. Адсорбция ионов на твердой поверхности. Понятие об ионитах. Обратимая ионообменная адсорбция – основа ионообменной хроматографии.
Физические процессы молекулярной адсорбции на твердой поверхности описываются уравнениями Ленгмюра и Фрейндлиха.
Уравнение Ленгмюра:
![]() ,
,
где Г – величина адсорбции, кмоль/кг или кмоль/м2 ;
Гmax – величина предельной адсорбции, кмоль/кг (кмоль/м2 );
С – концентрация раствора, кмоль/л;
а – константа равновесия адсорбции.
Это уравнение хорошо описывает адсорбцию для малых и больших концентраций растворов (или давлений газа).
Эмпирическое уравнение Фрейндлиха:
![]() ,
,
где Г – величина адсорбции, кмоль/кг (кмоль/м2 );
n – количество вещества-адсорбтива, кмоль;
m – масса адсорбента, кг;
К – константа (при С = 1 моль/л К = Г);
1/а – константа (адсорбционный показатель); зависит от природы адсорбента и температуры. 1/а = 0,1–1.
Адсорбционная хроматография основана на различии сорбируемости разделяемых веществ адсорбентом (твёрдое тело с развитой поверхностью); распределительная хроматография — на разной растворимости компонентов смеси в неподвижной фазе (высококипящая жидкость, нанесённая на твёрдый макропористый носитель) и элюенте (следует иметь в виду, что при распределительном механизме разделения на перемещение зон компонентов частичное влияние оказывает и адсорбционное взаимодействие анализируемых компонентов с твёрдым сорбентом).
Ионообменная хроматография основана на различии констант ионообменного равновесия между неподвижной фазой (ионитом) и компонентами разделяемой смеси.
Если на поверхности адсорбента уже адсобирован электролит, то при контакте этого адсорбента с другим электролитом почти всегда в той или иной степени происходит ионообменная адсорбция. Она наблюдается на поверхности с достаточно выраженным двойным электрическим слоем. Подвижные противоионы электрического слоя способны обмениваться на другие ионы того же знака, находящиеся в растворе.
Количественное описание ионообменного процесса (обратимость процесса, эквивалентность обмена, порядок обмена ионов) было сделано Гедройцем уже в начале XXв. Вещества, проявляющие способность к ионному обмену и используемые для адсорбции ионов, получили название ионообменников или ионитов.
Иониты имеют каркасную структуру, «сшитую» ковалентными связями. Каркас (матрица) обладает положительным или отрицательным зарядом, который скомпенсирован противоположным зарядом подвижных ионов – противоионов, находящихся в адсорбционной и диффузной частях двойного электрического слоя. Противоионы могут заменяться на другие ионы из раствора с зарядом того же знака, а каркас выступает в роли полииона и обусловливает нерастворимость ионита в растворителе.
Иониты делятся по составу на органические и неорганические, по происхождению – на природные и синтетические, по характеру обмениваемых ионов – на катиониты, аниониты и амфолиты.
Из природных неорганических катионитов чаще используются кристаллические силикаты типа цеолитов: шабазит, глакуонит и др. их каркас состоит из сетчатой структуры алюмосиликатов, в порах которой расположены ионы щелочных или щелочноземельных металлов, являющихся противоинами. К природным ионитам относятся апатиты.
Природные органические иониты – гумусовые вещества почв, содержащие карбоксильную группу, способную к ионному обмену. составляющие почву вещества обладают амфотерными свойствами и поэтому в зависимости от условий могут обменивать как катионы, так и анионы. Однако широкого применения природные иониты не имеют ввиду химической нестойкости и небольшой обменной емкости.
Промышленное применение имеют синтетические иониты, и среди них наиболее широко используют ионообменные смолы, которые имеют сетчатую структуру и содержат ионогенные группы: - OH, COOH, SO3 H, - COONa и т.п.
89. Написать формулу строения мицеллы золя, образованного в результате взаимодействия указанных веществ(избытка одного, затем другого вещества): CdCl 2 + Na 2 S ; FeCl 3 + NaOH . Назвать составляющие компоненты мицеллы.
1) CdCl2 + Na2 S
Избыток CdCl2 дает мицеллу:
[ (CdCl2 ) Cd2+ · Cl– ]+ x Cl–
![]()
зародыш: (CdCl2 )
![]()
ядро: [ (CdCl2 ) Cd2+
![]()
гранула: [ (CdCl2 ) Cd2+ · Cl– ]+
Избыток Na2 S дает мицеллу:
[2 (NaCl) 2 Cl– · Na+ ]– x Na+
зародыш: (NaCl )
![]()
ядро: (NaCl ) 2 Cl-
![]()
гранула: [ (CdCl2 ) Cd2+ · Cl– ]+
2) FeCl3 + NaOH
Избыток FeCl3 даетмицеллу:
[ (FeCl3 ) Fe3+ · 2Cl– ]+ x Cl–
зародыш: (FeCl3 )
ядро: (FeCl3 ) Fe3+
гранула: [ (FeCl3 ) Fe3+ · 2Cl– ]+
Избыток NaOH даетмицеллу:
[3 (NaCl) 3 Cl– · 2Na+ ]– x Na+
зародыш: (NaCl )
ядро: 3 (NaCl) 3 Cl–
гранула: [3 (NaCl) 3 Cl– · 2Na+ ]–
98. Коагуляция дисперсной системы. Скорость коагуляции. Причины, вызывающие процесс самопроизвольной коагуляции.
Коагуляция – процесс самопроизвольного укрупнения (слипания) дисперсных частиц, который может происходить при действии на дисперсную систему различных факторов: при интенсивном перемешивании или встряхивании, нагреве или охлаждении, облучении светом или пропускании электрического тока, при добавлении к системе электролитов или неэлектролитов и др. При разных способах воздействия на систему происходит уменьшение энергии связи диспергированных частиц с окружающей их дисперсионной средой. Так, добавление электролита вызывает сжатие диффузного слоя в коллоидной частице, следовательно, понижение величины электрокинетического потенциала. Это приводит к уменьшению электростатического отталкивания коллоидных частиц и, как следствие, к большей вероятности их слипания.
Минимальная концентрация электролита, добавляемого к дисперсной системе, при которой наступает явная коагуляция за определенный промежуток времени, носит название порога коагуляции ![]() . Порог коагуляции определяется температурой, природой добавленного электролита, знаком заряда добавляемого иона (действует прежде всего ион, имеющий заряд, противоположный заряду коллоидных частиц) и величиной заряда этого иона. Так, для трех-, двух- и однозарядных ионов явная коагуляция наступает при концентрации электролитов в соотношении 1:10 - 50: 500-1000 (приближенное правило Шульце-Гарди).
. Порог коагуляции определяется температурой, природой добавленного электролита, знаком заряда добавляемого иона (действует прежде всего ион, имеющий заряд, противоположный заряду коллоидных частиц) и величиной заряда этого иона. Так, для трех-, двух- и однозарядных ионов явная коагуляция наступает при концентрации электролитов в соотношении 1:10 - 50: 500-1000 (приближенное правило Шульце-Гарди).
Порог коагуляции рассчитывается следующим образом:
 , (1)
, (1)
где порог коагуляции, кмоль/м3 ;
С – молярная концентрация раствора электролита, кмоль/м3 ;
Vэл – объем раствора электролита, м3 ;
Vзоля – объем золя, м3 .
Теорию быстрой скорости коагуляции разработал Смолуховский. Он автор уравнения для расчета константы скорости коагуляции K:
 , (2)
, (2)
где n0
и n – число частиц в единице объема системы до начала коагуляции и к моменту времени ![]() соответственно
соответственно
время коагуляции, с.
Константа скорости коагуляции зависит от коэффициента диффузии для частиц и их радиуса следующим образом:
К = 8 · ·D · r.(3)
Учитывая уравнение (2) и уравнение Эйнштейна, окончательное уравнение для константы скорости коагуляции принимает вид:
 , (4)
, (4)
где К – константа скорости коагуляции, м3 /с;
![]() – вязкость среды, Па·с;
– вязкость среды, Па·с;
NA – число Авогадро.
Смолуховский ввел также понятие о времени половинной коагуляции, согласно которому время, необходимое для уменьшения первоначального числа частиц в 2 раза, связано с их исходным числом следующим образом:
 , (5)
, (5)
где – время половинной коагуляции, с;
– время от начала коагуляции, с.
Из уравнения, преобразованного к виду:
 , (6)
, (6)
следует, что если построенный в координатах nо /n = f ( график представляет собой прямую линию, то это служит показателем соответствия экспериментальных данных теории Смолуховского.
108. Суспензии. Условия их образования и свойства. Пасты – концентрированные суспензии. Примеры суспензий среди продуктов питания.
Суспензии – это взвеси порошков в жидкости (тип Т/Ж). Дисперсная фаза в суспензиях содержит частицы сравнительно больших размеров (более 10–4 см), поэтому суспензии седиментационно (т.е. по способности к оседанию) неустойчивы. Им не свойственны осмотическое давление, броуновское движение и диффузия. Частицы могут нести на своей поверхности двойной электрический слой, что способствует их стабилизации, но под влиянием электролитов суспензии коагулируют или образуют агрегаты, причем коагулированная суспензия, обычно легко пептизируется. Суспензии гидрофильных частиц стойки в воде, но нестойки в углеводородах. Их стойкость повышается в присутствии поверхностно-активных веществ. Повышению стабильности суспензий способствует также образование заряда на поверхности частиц (мицеллирование).
Пасты – это высококонцентрированные стабилизированные суспензии (типа Т/Ж), в которых частицы дисперсной фазы связаны за счет молекулярных сил и по этой причине не способны к взаимному перемещению. В таких высоковязких (пластично-вязких) системах почти вся дисперсионная среда сольватно связана с дисперсной фазой. Таким образом, пасты занимают промежуточное положение между порошками и суспензиями. В них могут протекать процессы, характерные для коллоидных систем с внутренней структурой (синерезис и др.) Большое практическое значение таких концентрированных систем обусловлено их пластичностью.
Примерами суспензий среди продуктов питания являются все пищевые пасты: томатная, шоколадная, сырная и т.д.
ЛИТЕРАТУРА
1. Ахметов Б. В. Задачи и упражнения по физической и коллоидной химии. – Л.: Химия, 1989.
2. Гамеева О. С. Физическая и коллоидная химия. – М.: Высшая школа, 1983.
3. Евстратова К. И., Купина Н. А., Малахова Е. М. Физическая и коллоидная химия. – М.: Высшая школа, 1990.
4. Зимон А. Д., Лещенко Н. Ф. Коллоидная химия. – М.: Химия, 2001.
5. Зимон А. Д., Лещенко Н. Ф. Физическая химия. – М.: Химия, 2000.
6. Киселев Е. В. Сборник примеров и задач по физической химии. – М.: Высшая школа, 1983.
7. Кнорре Д. Г. Физическая химия. – М.: Высшая школа, 1990.
8. Стромберг А. Г. Физическая химия. – М.: Высшая школа, 2001.
9. Степин Б. Д. Международные системы единиц физических величин в химии. – М.: Высшая школа, 1990.
10. Фридрихсберг Д. А. Курс коллоидной химии. – Л.: Химия, 1995.
11. Хмельницкий Р. А. Физическая и коллоидная химиия. – М.: Высшая школа, 1988.