| Скачать .docx |
Реферат: Теория Брэгга-Вильямса для неидеальных смесей
Реферат
по химии
"Теория Брэгга-Вильямса для неидеальных смесей"
2008
Свободная энергия смешения
Модель Брэгга-Вильямса иногда называют также теорией регулярных растворов. Она описывает жидкие смеси на основе простейших подходов статистической механики и не включает никаких сложных математических методов, кроме простой комбинаторики. Несмотря на свою простоту, теория дает удивительно хорошее качественное описание множества очень сложных процессов в жидких смесях. Эта модель лежит в основе теории растворов полимеров Флори-Хаггинса. Некоторые концепции, например параметр ч, введенный в модели Брэгга-Вильямса, используются в различных ситуациях, поэтому важно знать их происхождение.
Модель Брэгга-Вильямса основана на решеточной модели, в которой каждая позиция решетки может разместить одну молекулу независимо от ее типа и размера. В таком случае число соседей всегда постоянно, если считать, что все места в решетке заняты и что объем не меняется при смешении. Основные постулаты модели сводятся к следующему.
1. Компоненты смеси смешиваются хаотически.
2. Число соседних молекул постоянно.
3. Взаимодействие ограничивается ближайшими соседями.
Из этих постулатов следует, что энергия смешения будет ненулевой, если мы предположим, что энтропия смешения идеальна AS = Аидеал . Это приближение среднего поля обсуждается ниже.
Рассмотрим смешение двух веществ А и В.
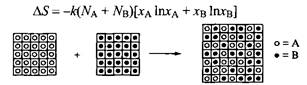
Рис. 1. Решеточная модель хаотического смешения двух жидкостей
Энтальпия смешения АН рассчитывается как разность энергий взаимодействия между молекулами двух типов. Полные энергии чистых индивидуальных компонентов равны
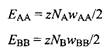
Полная энергия смеси записывается как
![]()
Таким образом, получаем:
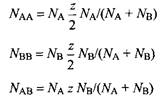
Изменение внутренней энергии при смешении равно энергии смеси за вычетом энергии двух индивидуальных жидкостей:
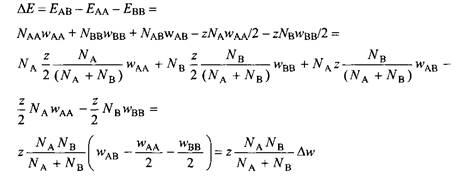
В уравнении введена величина Aw, равная
![]()
Видно, что в уравнение входит только величина Днн, и результат в неявном виде зависит от параметров индивидуальных взаимодействий waa, wbb и wab- Изменение взаимодействий при смешении, очевидно, может быть как положительным, так и отрицательным. Знак изменения зависит от того, является ли взаимодействие АВ более положительным по сравнению с усредненными взаимодействиями А А и ВВ.
В решеточную модель не входит член, зависящий от давления и объема, поэтому изменение энтальпии можно записать как
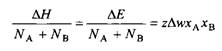
Наконец, вводя параметр взаимодействия ч, определяемый согласно соотношению
![]()
найдем выражение для энтальпии смешения в расчете на один моль вещества:
![]()
Теперь нужно получить выражение для важнейшей величины — свободной энергии смешения в расчете на моль вещества:
![]()
Из выражения для свободной энергии можно рассчитать целый ряд важных величин. Например, химический потенциал компонента А в смеси описывается выражением
![]()
Следует отметить, что уравнение, полученное очень простым способом, представляет выражение для химического потенциала неидеальной смеси. Последний член в нем — это интересующая нас избыточная величина:
![]()
Таким образом, химический потенциал компонента А в смеси записывается в следующем виде:
![]()
где ад — активность; а коэффициент активности компонента А можно определить следующим образом:
![]()
Исторически параметр ч сначала рассматривался как энтальпийная величина, как в уравнении. Позднее параметр ч был идентифицирован как величина свободной энергии, что подтверждается уравнением.
Закон Рауля
Давление пара над идеальным раствором описывается законом Рауля![]() Для неидеального раствора закон Рауля записывается с учетом активности
Для неидеального раствора закон Рауля записывается с учетом активности ![]() Следуя модели Брэгга-Вильямса и используя уравнение, получим:
Следуя модели Брэгга-Вильямса и используя уравнение, получим:
![]()
Параметр взаимодействия![]() может быть положительным или отрицательным, что проявляется как положительное или отрицательное отклонение от закона Рауля.
может быть положительным или отрицательным, что проявляется как положительное или отрицательное отклонение от закона Рауля.
Фазовое разделение
Для того чтобы жидкости смешивались, необходимо выполнение условия AG < 0. Но этого условия недостаточно для полного смешения жидкостей при любых концентрациях. Изменение свободной энергии при смешении, отнесенное к одному молю, определяется выражением
![]()
На рис. 3 представлены зависимости изменения свободной энергии при смешении ) от мольной доли компонента А для разных значений параметра ч. Верхняя кривая соответствует разделению на фазы при начальной мольной доле 0.5. При этом образуются две фазы, концентрации которых отвечают точкам end.
Уравнение предсказывает, что разделение смеси на фазы происходит всегда прежде всего при условии jca = *в· Разделению смеси на фазы отвечает выполнение условия![]()
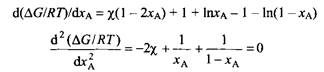
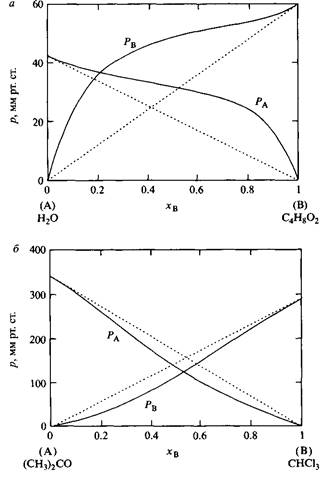
Рис. 2. Вода и диоксан ведут себя как несмешивающиеся жидкости, ацетон и хлороформ, напротив, смешиваются между собой. © 1962 Prentice-Hall Inc., Englewood Cliffs, NJ. С разрешения PearsonEducationLimited
Из уравнения следует, что ч = 2 приход*в = 0.5, т. е. разделение смеси на фазы будет происходить при ч > 2. Температура, при которой начинается фазовое разделение, называется критической температурой:
![]()
Теория Брэгга-Вильямса отражает основы физики фазового разделения простых двухкомпонентных систем. Однако известны смеси двух жидкостей, которые полностью смешиваются при низких температурах, но разделяются на фазы при повышении температуры. На рис. 10.4 показаны различные случаи фазового поведения смесей. При повышенных температурах все системы неизбежно обнаруживают верхнюю критическую точку. Это соответствует замкнутой области смешения.
Считается, что нижняя критическая точка связана с влиянием температуры на изменение взаимодействий в смеси Aw. Но эти представления выходят за рамки модели Брэгга-Вильямса и не вносят ничего нового в понимание физического смысла явления. Для систем полиэтиленоксид вода нижняя критическая точка, возможно, появляется вследствие индуцированных температурой структурных изменений оксиэтиленовых групп. Другими словами, для таких систем важную роль играют внутренние степени свободы, что не рассматривает модель Брэгга-Вильямса.
Полезно рассмотреть смешение воды и гидрофобных молекул, например углеводородов. Взаимодействие вода-вода отрицательно и существенно по величине, тогда как взаимодействия вода-углеводород и углеводород-углеводород по сравнению с ww слабые и их можно считать нулевыми. Тогда эффективное парное взаимодействие записывается следующим образом:
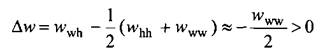
Параметр ч для такой системы при комнатной температуре будет иметь значение, близкое к 5. Таким образом, теория Брэгга-Вильямса предсказывает несмешиваемость масла и воды.
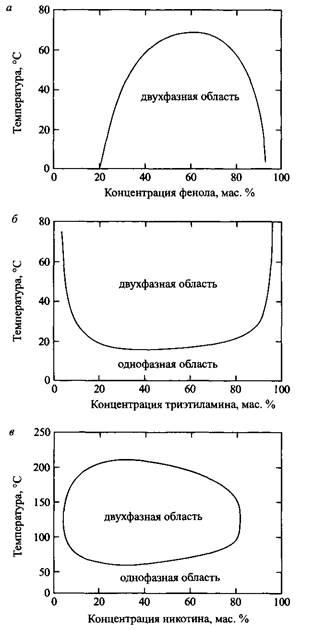
Рис. 3. Фазовые диаграммы смесей с верхними и нижними критическими точками: а — фенол-вода; б — триэтиламин-вода; в — никотин-вода
Теория растворов полимеров Флори-Хаггинса: описание фазовых превращений
Энтропия смешения для системы полимер-растворитель
Рассмотрим смесь двух жидкостей, одна из которых — полимер. При этом можно использовать приведенную выше модель, но энтропия смешения для системы растворитель-полимер будет другой. Очевидно, что изменение энтропии будет меньше, поскольку мономерные звенья полимера не способны полностью использовать увеличение объема при смешении. Этому препятствует «связанность» мономеров в полимере. А энергия смешения имеет тот же вид, что и для смеси двух низкомолекулярных жидкостей.
Рассмотрим раствор, состоящий изН\молекул растворителя и N2 молекул полимера со степенью полимеризации г; суммарно число молекул растворителя и мономерных звеньев равноН= N + Nir. Энтропия смешения такой системы выражается уравнением
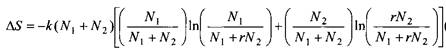
где![]()
![]()
Тогда выражение для энтропии смешения можно записать в обычном виде:
![]()
Общее число молей определяется как число молей растворителя и полимерных сегментов в системе. Энтропия смешения, отнесенная к одному молю вещества, задается выражением
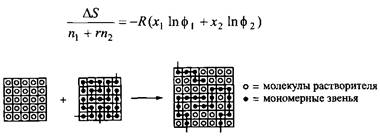
Рис. 4. Модель решетки для случайного смешения полимера и жидкости светлые кружочки)
Энергия смешения имеет ту же форму, что и для смеси низкомолекулярных жидкостей); запишем ее в виде
![]()
Интересно проанализировать разницу в изменении энтропии при смешении двух простых жидкостейи смешении простой жидкости и полимера. Обозначим эту разницу AAS:
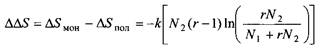
Таким образом, величина AASувеличивается с длиной молекулы полимера, и, как следствие, можно ожидать, что фазовое разделение смеси полимер-растворитель должно происходить на более ранней стадии, т. е. при более низкой температуре, чем фазовое разделение смесей двух низкомолекулярных жидкостей.
Фазовое равновесие в теории Флори-Хаггинса
Из уравнений и свободную энергию смешения на моль можно выразить уравнением, где первый член представляет собой изменение энергии, а второй — изменение энтропии при смешении. Производная этого выражения по компоненту 1 соответствует химическому потенциалу растворителя в бинарном растворе:
![]()
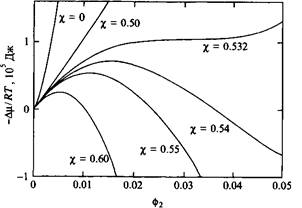
Рис. 5. Избыточный химический потенциал растворителя в бинарном растворе, содержащем полимер. Степень полимеризации составляет 1000, значения параметра взаимодействия ч указаны около кривых
![]()
Немонотонное изменение химического потенциала свидетельствует о фазовом разделении системы. Интересно выяснить, при каком значении ц это происходит. После некоторых алгебраических преобразований найдем, что критическая точка определяется двумя выражениями:
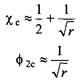
Имеет смысл сравнить значения критических параметров для растворов полимеров с соответствующими параметрами для регулярных растворов хс = 2ифс = 0.5. Видно, что растворы полимеров легче становятся несовместимыми и разделяются на фазы ).
И-Температура
В науке о полимерах широко распространена концепция и-температуры и представление о хороших и плохих растворителях. Чтобы ввести эти понятия, вернемся к уравнению. Избыточный химический потенциал для малых объемных долей растворенного вещества можно разложить в ряд:
![]()
где тета-температура определяется как
![]()
Уравнение показывает, что при равенстве физической температуры тета-температуре система ведет себя как идеальный раствор, т. е. Дмй = 0. Если Ф >и, растворитель является для полимера хорошим растворителем, а если Ф <и — растворитель плохой. Кроме того, можно интерпретировать 0-темпера-туру другим способом, используя критическую температуру, при которой наблюдается первое фазовое разделение раствора полимера:
![]()
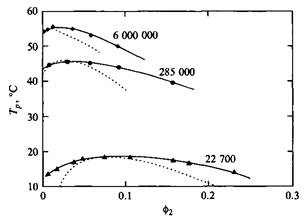
Рис. 6. Фазовые диаграммы для трех фракций полиизобутилена в диизобутилкетоне. Сплошные кривые проведены через экспериментальные точки, пунктирные кривые — теоретические
Таким образом, из уравнения и-температуру можно определить как критическую температуру для бесконечно длинного полимера.