| Скачать .docx |
Реферат: Теория химико-технологических процессов
Федеральное агентство по образованию РФ
Государственное образовательное учреждение высшего профессионального образования
Волгоградский государственный технический университет
(ВолгГТУ)
Кафедра «Технология органического и нефтехимического синтеза»
Семестровая работа
по курсу:
«Теория химико-технологических процессов»
Выполнил:
Проверил:
Оценка работы ____________ баллов.
Волгоград 2008г.
Содержание:
Введение ……………………………………..…………………………….... 3
1. Задание ……………………………………………………………………….. 4
2. Литературный обзор ……….………………………………………………... 5
2.1. Гипотеза о схеме превращения …….…………………………………... 5
2.2. Гипотеза о механизме реакции …………………………………………. 5
2.2.1. Влияние на протекание реакции атакующей частицы ………….. 5
2.2.2. Влияние на протекание реакции строения субстрата …………... 6
2.2.3. Влияние на протекание реакции строение растворителя ………. 8
2.2.4. Влияние на протекание реакции строение уходящей частицы .. 10
3. Обсуждение результатов …………………………………………………... 12
3.1. План кинетических экспериментов …………………………………... 12
3.2. Анализ первичных кинетических кривых …………………………… 14
3.3. Расчёт параметров кинетической модели ……………………………. 15
3.4. Проверка адекватности кинетической модели эксперименту ……… 24
4. Экспериментальная часть ………………………………………………….. 26
4.1. Выбор метода анализа ключевого компонента ……………………… 26
4.2. Приготовление растворов реагирующих веществ …………………... 39
4.3. Схема установки, подбор реактора и доказательство его идеальности ……………………………………………………………………………. 40
4.4. Прописи кинетических экспериментов ……………………………… 43
Выводы …………………………………………………………………….. 47
Список используемой литературы ……………………………………….. 47
Введение
Современная химическая промышленность выпускает огромное количество разнообразнейших продуктов и товаров, что естественно связанно с проведением, оптимизацией и управлением определённых процессов, в частности химических реакций. Это требует проведения огромной научно-исследовательской работы, позволяющей переносить химические реакций, необходимых для получения ценных химических продуктов из лабораторных условий на промышленный уровень.
А поскольку научно-исследовательская работа является неотъемлемой частью научно-технического прогресса, то это говорит о её значимости. Она является постоянным притоком всё новых и новых знаний о природе вещества в общемировую копилку знаний человечества. Полезность научно-исследовательской работы в области изучения химической технологии неоценима, поскольку на данный момент продукты химической промышленности присутствуют абсолютно во всех сферах нашей жизнедеятельности, и не могут быть никоим образом изъяты.
Как и везде, в научно-исследовательской работе присутствуют проблемы, и главная из них – это динамика развития научно-технического прогресса, она постоянно растёт, и требует сокращения времени отводимого на исследования. Причём основной проблема исследований химических реакций является изучения механизма реакции, что процесс очень трудоёмкий и дорогостоящий. Что служит главным препятствием запуска процесса в производство. Поэтому задачу ставят по-другому - оптимизация и управление процессом, не зная его механизма. Это осуществляется путём исследования системы по изменению значений скорости реакции (функций отклика) при воздействии на систему каким-то фактором, уровень которого известен, т.е. изучение кинетики реакции.
Поэтому целью данной работы является приобретение знаний и умений, а также первоначальных навыков в изучении и применении кинетических исследований химических реакций, в частности построение кинетической модели реакции, т.е. отыскание вида математических уравнений, описывающих скорость изменения концентрации веществ во времени, и нахождение значений параметров, входящих в эти уравнения. В частности, построение кинетической модели реакции, т.е. отыскание вида математических уравнений, описывающих скорость изменения концентрации веществ во времени, и нахождение значений параметров, входящих в эти уравнения.
Для достижения цели необходимо решить следующие задачи: произвести анализ литературных данных, определиться и провести экспериментальную часть работы и обсудить результаты, итогом которых должна стать кинетическая модель химического процесса.
1. Задание
| № варианта | Реагенты | Температурный интервал | Растворитель | ||
| A | Y | Ключевой | |||
| 65 | CH3 Cl | KI | Y | 80-110 | водный раствор спирта |
2. Литературный обзор
2.1. Гипотеза о схеме превращений.
Основная реакция:
CH3 Cl + KJ → CH3 J + KCl
Побочные реакции:
CH3 Cl + KJ → CH2 JCl + KH СH3 I + KCl → CH3 Cl + KI
R-OH + KJ → R-J + KOH
2.2. Гипотеза о механизме реакции.
Предположительным механизмом реакции является бимолекулярное нуклеофильное замещение SN 2 .
Вначале предполагается образование нуклеофильной частицы в результате диссоциации соли:
KJ → K+ + J-
Далее ион йода атакует атом углерода, имеющий частичный положительный заряд, со стороны, противоположной атому хлора, т. е. «с тыла». Образование связи С-J и разрыв связи С-Cl в переходном состоянии происходят одновременно.
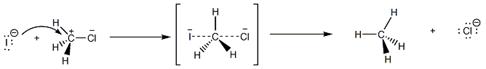
Переходное состояние можно представить как активированный комплекс, в котором атом углерода частично связан со вступающей группой, а связь С-Cl с уходящей группой еще не вполне разорвалась. Отрицательный заряд распределен между вступающей и уходящей группами. При этом атом углерода остается незаряженным. Таким образом, реакция протекает как одностадийный синхронный процесс.
Следует отметить, что скорость реакции SN 2 подчиняется кинетическому уравнению второго порядка:
w = k2 [CH3 Cl] [J- ],
т. е. пропорциональна концентрации субстрата и нуклеофила [4].
2.2.1. Влияние на протекание реакции атакующей частицы.
Нуклеофильность нейтральной молекулы или отрицательно заряженной частицы определяется способностью отдавать пару электронов при образовании связи с любым атомом, кроме водорода.
Нуклеофилы значительно чувствительны к стерическим затруднениям реакции. Способность нуклеофила участвовать в нуклеофильной атаке зависит и от характера его ВЗМО. Наибольшая нуклеофильность характерна для атомов и частиц, имеющих электроны на несвязывающих орбиталях.
Анионы являются значительно более сильными нуклеофилами, чем сопряженные им кислоты:
![]()
В общем, нуклеофильность зависит от электроотрицательности атома и от растворителя. По периоду слева направо нуклеофильность уменьшаются:
![]()
В протонных растворителях по группе сверху вниз нуклеофильность возрастает:
![]()
Если нуклеофильным центром в ряду частиц выступает один и тот же атом, нуклеофильность этих частиц изменяется параллельно изменению их основности:
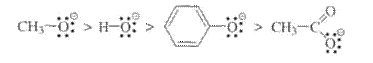
Оценки степени нуклеофильности различных нуклеофильных реагентов обобщены в таблице:
| Степень нуклеофильности | Нуклеофилы | Относительная активность |
| Очень сильные нуклеофилы | I- , HS- , RS- | >105 |
| Сильные нуклеофилы | Br- , HO- , RO- , CN- , N3 - | 104 |
| Умеренные нуклеофилы | NH3 , Cl- , F- , RCOO- | 103 |
| Слабые нуклеофилы | H2 O, RON | 1 |
| Очень слабые нуклеофилы | RCOOH | 10-2 |
Как видно из этой таблицы, очень сильными нуклеофилами являются частицы, в которых нуклеофильным центром выступают атомы третьего или более высоких периодов (сера, фосфор, йод) [4].
2.2.2. Влияние на протекание реакции строение субстрата.
Для того чтобы выяснить возможность перехода от одного механизма реакции к другому, необходимо рассмотреть влияние как электронных, так и стерических факторов на переходное состояние. В случае, если атака осуществляется по механизму SN 2 , можно ожидать, что возрастание индуктивного эффекта по мере увеличения числа метальных групп должно приводить к постепенному уменьшению положительного заряда на атоме углерода, связанном с галогеном, и, следовательно, к затруднению атаки этого атома нуклеофилом.
Этот эффект выражен не очень сильно, вследствие чего наиболее важную роль должны играть стерические факторы. Этим можно объяснить тот факт, что по мере увеличения объема заместителей у атома углерода, связанного с галогеном, возможность атаки этого атома углерода нуклеофилами существенно затрудняется. Кроме того, следует иметь в виду, что при атаке по механизму SN 2 этот атом углерода окружен в переходном состоянии пятью группами (в отличие от молекул исходного галогенида, в которых его окружают только четыре группы), поэтому чем больше объем трех заместителей в исходном галогениде (не считая атома галогена), тем им должно быть более тесно в переходном состоянии и тем, следовательно, неохотнее будет происходить образование этого состояния.
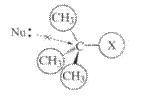
Очевидно, что плоскостное расположение стерически наиболее выгодно, поскольку три заместителя располагаются при этом на максимальных расстояниях один от другого (ср. с четырьмя заместителями в исходном галогениде и с пятью заместителями в переходном состоянии при механизме SN 2 ). Понятно, что преимущество такого расположения будет проявляться тем в большей степени, чем больше объем заместителей.
В зависимости от строения субстрата реакционная способность галогеналканов в реакциях SN 2 поэтому уменьшается в ряду:
галогенметаны > первичные галогеналканы > вторичные галогеналканы >> третичные галогеналканы
Реакции SN 2 с участием аллил- и бензилгалогенидов ускоряются вследствие стабилизации переходного состояния, достигаемой сопряжением π-связи (или π-электронного облака бензольного кольца) с p-орбиталью, формирующейся на атоме углерода в переходном состоянии. Этот эффект сопряжения понижает энергию активации реакции SN 2 и тем самым увеличивает ее скорость.
Очень интересным примером влияния строения на реакционную способность галогенпроизводных может служить 1-бром-триптицен II :
Атом брома в этом соединении совершенно инертен по отношению к действию нуклеофильных агентов. Атака «сзади» по механизму SN 2 затруднена стерическими факторами, поскольку молекула имеет форму клетки, а для образования переходного состояния, в котором с атомом углерода должны быть связаны три замещающие группы, требуется, чтобы эти группы прошли через копланарное расположение, что невозможно, так как атом углерода жестко связан со стоящими при нем заместителями.
Ниже даны количественные оценки относительной реакционной способности различных галогеналканов в реакциях вида:
![]()
Относительные скорости реакций SN 2 галогеналканов R-X [4]:
| Заместитель R | CH3 | C2 H5 | (CH3 )2 CH | (CH3 )3 C | CH2 =CHCH2 |
| Относительная скорость | 30 | 1 | 0,03 | ~ 0 | 50 |
2.2.3. Влияние на протекание реакции строение растворителя.
Реакции SN 2 , как правило, проводят в растворителях. Присутствие всех реагирующих веществ в гомогенной системе способствует протеканию реакции. Растворители классифицируют по ионизирующей силе, т. е. по способности разделять молекулы на ионы. Различают неполярные, полярные протонные и полярные апротонные растворители. Примеры этих растворителей приведены ниже. Оценивая свойства растворителей, следует иметь в виду, что ионизирующая способность растворителей изменяется соответственно изменению диэлектрической проницаемости (ε).
Неполярные растворители не растворяют основания и соли, которые часто выступают в роли нуклеофильных реагентов. Эти растворители в реакциях нуклеофильного замещения применяют редко.
| Растворитель | ε |
| Алканы, петролейный эфир | < 2 |
| CCl4 | 2,23 |
| C6 H6 | 2,28 |
| CHCl3 | 4,7 |
Солеобразные нуклеофильные реагенты лучше всего растворяются в воде, которая является полярным протонным растворителем . Однако в воде плохо растворяется большинство органических соединений, в том числе галогенпроизводные. Поэтому часто применяют смеси растворителей (водный этанол, водный диоксан, водный ацетон).
| Растворитель | ε |
| CH3 COOH | 6,2 |
| NH3 (жидк.) | 17 |
| C2 H5 OH | 24 |
| CH3 OH | 32,6 |
| HO(CH2 )2 OH | 37,7 |
| HCOOH | 59 |
| H2 O | 80 |
Эффективными являются также полярные апротонные растворители , как правило, имеющие умеренную диэлектрическую проницаемость, но не способные к образованию водородных связей.
| Растворитель | ε |
Диоксан
|
2,2 |
Диэтиловый эфир
|
4,34 |
Тетрагидрофуран
|
7,4 |
Ацетон
|
25 |
Диметилформамид (ДМФА)
|
38 |
Диметилсульфоксид (ДМСО)
|
46 |
Влияние растворителя на скорость реакций SN 2 зависит от характера нуклеофильного реагента (нейтральная молекула или анион), который определяет тип переходного состояния и, в частности, степень разделения зарядов в нем по сравнению с исходными реагентами:
а) нуклеофильный реагент - нейтральная молекула.
![]()
Переходное состояние в такой реакции более полярно, чем исходные соединения. Увеличение полярности растворителя приводит к лучшей сольватации переходного состояния и, следовательно, к росту скорости реакции. Эти реакции ускоряются полярными растворителями, как протонными (спирты, карбоновые кислоты), так и апротонными (ацетон, диметилсульфоксид).
Реакции алкилирования аминов представляют собой типичный пример реакций SN 2 , в которых в качестве нуклеофила выступают нейтральные молекулы. Такие реакции гладко протекают и в спиртах.
б) нуклеофильный реагент – анион.
Переходное состояние этой реакции менее полярно, чем исходные реагенты, поскольку в нем отрицательный заряд распределяется между входящим нуклеофилом и уходящим галогеном. Изменения в сольватирующих свойствах растворителей в таких реакциях сказываются прежде всего на активности нуклеофила.
Полярные протонные растворители за счет водородных связей сольватируют нуклеофил-анион и понижают тем самым его энергию и реакционную способность.
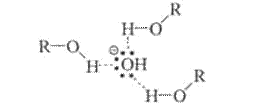
Полярные апротонные растворители, многие из которых хорошо растворяют как соли, так и органические соединения, наиболее часто применяют в реакциях SN 2 . Молекулы полярных апротонных растворителей способны специфически сольватировать катионы и не могут сольватировать анионы. Многие реакции SN 2 проходят в таких растворителях на несколько порядков быстрее, чем в протонных.
В качестве примера нуклеофильного замещения с участием заряженного нуклеофила можно указать на реакции гидролиза и алкоголиза галогеналканов [4].
2.2.4. Влияние на протекание реакции строение уходящей частицы.
В ряду связей: C-F, C-Cl, C-Br, C-I реакционная способность в реакциях SN 2 возрастает, поскольку в этом ряду увеличиваются длина связи С-Х, ее поляризуемость, а также устойчивость уходящей группы - галогенид-иона.
Чем выше основность отщепляемой группы, тем труднее она отщепляется атакуемым нуклеофилом. Такие сильно основные группы, как, например, -OR, -OH, -NH2, связанные с углеродом через небольшие трудно поляризуемые атомы, в обычных условиях, как правило, не способны к замещению, однако в кислой среде они все же могут замещаться вследствие начальной протонизации, приводящей к образованию положительно заряженных частиц (а не нейтральных молекул), атакуемых нуклеофилом; в результате замещение менее основной группировки YH может идти легче, чем аниона Y- , хотя последний и является более сильным основанием.
Именно этим можно объяснить тот факт, что даже крайне прочно удерживаемый атом фтора в алкилфторидах может замещаться нуклеофильными реагентами в концентрированном растворе серной кислоты. И использование иодистого водорода для расщепления эфиров основано на том обстоятельстве, что ион I- представляет собой наиболее мощный нуклеофильный реагент, какой может быть получен в растворе сильной кислоты, которая, следовательно, необходима для реализации этой реакции.
Выше уже отмечалось, что перечень субстратов в реакциях SN 2 не ограничивается галогеналканами. В качестве реакционноспособных субстратов в них выступают, в частности, эфиры сульфокислот.
![]()
При этом группы RSO2 O- оказываются лучшими уходящими группами, поскольку являются сопряженными основаниями очень сильных алкил- или арилсульфоновых кислот.
По аналогичным причинам наиболее плохие уходящие группы - гидроксид-ион - ОН и амид-ион - NH2. Будучи сопряженными основаниями очень слабых кислот, эти ионы отличаются высокими энергиями образования и крайне редко выступают в качестве уходящих групп.
Ниже перечислены различные уходящие группы в ряду снижения легкости их отщепления (или снижения их «качества») [4]:
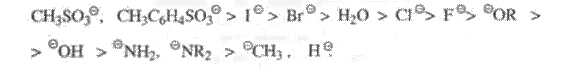
3. Обсуждение результатов
3.1. План кинетических экспериментов.
Анализ литературных данных, проведённых по учебной литературе, позволил предположить, что изучаемая реакция протекает, вероятнее всего, по механизму бимолекулярного нуклеофильного замещения (SN 2 ).
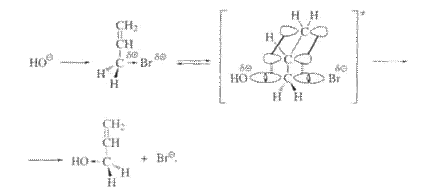
Схема механизма реакции:
![]()
Кинетическое уравнение для данного механизма:
![]() (3.1)
(3.1)
Оно включает неизмеряемую концентрацию переходного состояния С X - . Из схемы механизма реакции видно:
![]() (3.2)
(3.2)
Воспользовавшись принципом стационарности Боденштейна – Семёнова [3], в соответствии с которым в установившемся режиме:
![]() (3.3)
(3.3)
Отсюда получаем:
![]() (3.4)
(3.4)
Откуда найдём выражение для С X - :
![]() (3.5)
(3.5)
При подстановке этого выражения в уравнение (3.1) получим кинетическое уравнение для механизма SN 2 :
![]() (3.6)
(3.6)
Уравнение (3.6) включает 3 константы скорости: k1 и k2 – константы скоростей первой и второй реакции, и k-1 – константу скорости реакции распада промежуточной частицы на первой стадии реакции. При экспериментальном исследовании реакции необходимо решить вопрос о вкладе процесса распада промежуточного комплекса. Если эта реакция вносит существенный вклад в процесс, то кинетическая модель реакции будет представлена уравнением (3.6). Если же вклад обратной реакции мал, т.е. r-1 << r2 , уравнение (3.6) значительно упрощается, т.к. в знаменателе можно пренебречь слагаемыми k-1 , что приводит уравнение к виду (3.7):
![]() (3.7)
(3.7)
Таким образом, формируется план эксперимента, в котором необходимо предусмотреть опыты по выявлению вклада обратной реакции, чувствительности скорости реакции к изменению реакции концентрации каждого реагента, изучение влияния температуры на скорость реакции. В план эксперимента включены также опыты для проверки адекватности полученной модели.
Для рассматриваемой реакции можно предложить следующий план эксперимента:
Таблица 3.1. План эксперимента.
| Номер опыта | Начальные концентрации, моль/л | Темпера-тура, ºC | СZ , моль/л |
СY , моль/л |
Назначение опыта | ||
| CA,O | CY,O ключ. компонент | CZ,O | |||||
| 1 | 1,0 | 0,5 | 0 | 80 | 0,4 | 0,1 | Базовый опыт |
| 2 | 0,9 | 0,5 | 0 | 80 | 0,4 | 0,1 | Влияние концентрации компонента A на скорость реакции |
| 3 | 1,0 | 0,4 | 0 | 80 | 0,32 | 0,08 | Влияние концентрации компонента Y на скорость реакции |
| 4 | 1,0 | 0,5 | 0,2 | 80 | 0,6 | 0,1 | Вклад обратной реакции |
| 5 | 1,0 | 0,5 | 0 | 80 | 0,4 | 0,1 | Опыты на воспроизводимость |
| 6 | 1,0 | 0,5 | 0 | 80 | 0,4 | 0,1 | |
| 7 | 1,0 | 0,5 | 0 | 80 | 0,4 | 0,1 | Изучение влияния температуры на скорость реакции |
| 8 | 1,0 | 0,5 | 0 | 90 | 0,4 | 0,1 | |
| 9 | 1,0 | 0,5 | 0 | 100 | 0,4 | 0,1 | |
| 10 | 0,95 | 0,4 | 0,125 | 110 | 0,445 | 0,08 | Проверка адекватности |
3.2. Анализ первичных кинетических кривых.
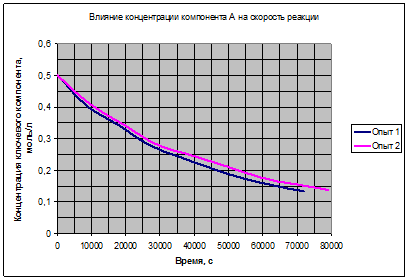
Рис. 3.1. Влияние компонента Aна скорость реакции.
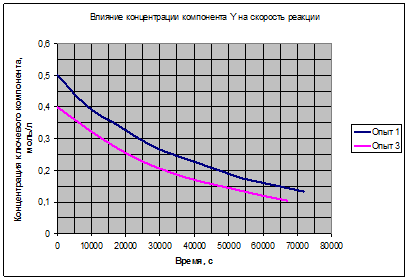
Рис. 3.2. Влияние концентрации Yна скорость реакции.
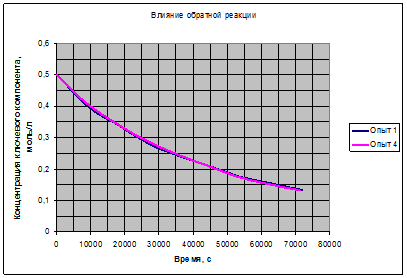
Рис. 3.3. Вклад обратной реакции.
3.3. Расчёт параметров кинетической модели.
Полученные зависимости на рис. 3.1, 3.2, 3.3 подтверждают возможность выбора для расчёта уравнения (3.7). Таким образом, можно считать основной гипотезой кинетической модели данной реакции выражение (3.7):
![]() (3.7)
(3.7)
Поиск констант можно осуществить интегральным и дифференциальными методами обработки результатов [?]. Воспользуемся интегральным методом, суть которого сводиться к поиску констант для интегрального решения уравнения. Для уравнения (3.7) интегральное решение имеет вид (3.8) [2]:
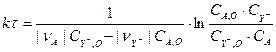 (3.8)
(3.8)
Где νA , νY - - стехиометрический коэффициент для вещества Aи Y- соответственно. А т.к. в стехиометрическом уравнении реакции νA = 1, νY - = 1, отсюда выражение (3.8) принимает следующий вид:
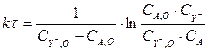 (3.9)
(3.9)
Где CA , O , С Y -, O – начальная концентрация вещества Aи Y- соответственно. Т.к. в уравнение (3.9) входит не определяемая в опыте текущая концентрация CA , её необходимо выразить через измеряемые концентрации CA , O , С Y -, O , С Y - , это возможно из уравнения материального баланса:
![]() (3.10)
(3.10)
Подставляя это выражение в уравнение (3.9), получим:
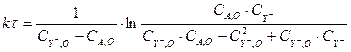 (3.11)
(3.11)
Для отыскания констант воспользуемся линейным методом наименьших квадратов [?]. Используя метод функциональных шкал, можно провести линеаризацию уравнения (3.11). Для этого положим:
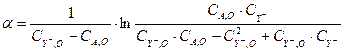 (3.12)
(3.12)
Тогда зависимость (3.11) запишется в виде линейной функции:
![]() (3.13)
(3.13)
3.3.1. Расчёт кинетической модели базового опыта.
При температуре t=80ºС, СA, O =1,0 моль/л, СY -, O =0,5 моль/л, τ1 = 9000, τ2 = 18000, …, τ8 =72000 были получены такие данные:
Таблица 3.2. Результаты базового опыта, приведённые для линейной функции (3.11).
| i | τi | 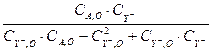 |
αi | |
| 1 | 9000 | 0,9 | 0,888889 | 0,235566 |
| 2 | 18000 | 0,841 | 0,810939 | 0,419124 |
| 3 | 27000 | 0,78 | 0,717949 | 0,662714 |
| 4 | 36000 | 0,741 | 0,650472 | 0,860113 |
| 5 | 45000 | 0,707 | 0,585573 | 1,070329 |
| 6 | 54000 | 0,674 | 0,51632 | 1,322055 |
| 7 | 63000 | 0,653 | 0,468606 | 1,515984 |
| 8 | 72000 | 0,633 | 0,420221 | 1,733948 |
Для функции (3.13) можно оценить коэффициент kпо формуле [1]:
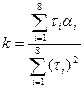 (3.14)
(3.14)
Для этого составим расчётную таблицу 3.3:
Таблица 3.3. Расчётная таблица для базового опыта.
| i | τi | αi | (τi )2 | τi αi |
| 1 | 9000 | 0,235566 | 81000000 | 2120,095 |
| 2 | 18000 | 0,419124 | 324000000 | 7544,232 |
| 3 | 27000 | 0,662714 | 729000000 | 17893,29 |
| 4 | 36000 | 0,860113 | 1296000000 | 30964,07 |
| 5 | 45000 | 1,070329 | 2025000000 | 48164,82 |
| 6 | 54000 | 1,322055 | 2916000000 | 71390,98 |
| 7 | 63000 | 1,515984 | 3969000000 | 95507 |
| 8 | 72000 | 1,733948 | 5184000000 | 124844,3 |
| Сумма: | - | - | 16524000000 | 398428,8 |
Отсюда по формуле (3.14):
![]()
3.3.2. Оценка дисперсии воспроизводимости.
Её можно произвести по двум параллельным выборкам (опытам 5, 6) с учетом того, что в выборках одинаковое число членов по следующей формуле (3.15) [1]:
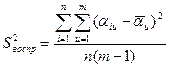 (3.15)
(3.15)
Где ![]() - ошибка опыта,
- ошибка опыта, 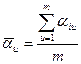 , n – количество членов параллельной выборки, m – количество параллельных опытов. При этом число степеней свободы равно
, n – количество членов параллельной выборки, m – количество параллельных опытов. При этом число степеней свободы равно ![]() .
.
Т.к. изучается единый технологический процесс, протекающий на одной и той же установке, и так как нет резко выделяющихся значений, то не будем проверять однородность и нормальность результатов параллельных опытов, используя статистику.
Проверка осуществляется по общей схеме проверки гипотез. Для условий базового опыта (t=80ºС, СA, O =1,0 моль/л, СY -, O =0,5 моль/л), при n = 8, т.е. τ1 = 8900, τ2 = 17800, …, τ8 =71200, и m = 2 (число параллельных опытов) получены следующие данные и из них получены выражения для расчета дисперсии воспроизводимости. Все они сведены в таблице 3.4.
Таблица 3.4. Расчётная таблица для дисперсии воспроизводимости базового опыта.
| i | τi | Опыт 5 | Опыт 6 | Опыт 5 | Опыт 6 | |||||
| CY -,i | αi | CY -,i | αi | |||||||
| 1 | 8900 | 0,406 | 0,219078 | 0,403 | 0,227278 | 0,223178 | 0,0041 | 1,68∙10-5 | 0,0041 | 1,68∙10-5 |
| 2 | 17800 | 0,336 | 0,436741 | 0,343 | 0,412179 | 0,42446 | 0,0122 | 0,000151 | 0,0122 | 0,000151 |
| 3 | 26700 | 0,281 | 0,658147 | 0,282 | 0,653601 | 0,655874 | 0,0022 | 5,16∙10-6 | 0,0022 | 5,16∙10-6 |
| 4 | 35600 | 0,242 | 0,854529 | 0,243 | 0,848975 | 0,851752 | 0,0027 | 7,71∙10-6 | 0,0027 | 7,71∙10-6 |
| 5 | 44500 | 0,205 | 1,084081 | 0,208 | 1,063518 | 1,073799 | 0,0102 | 0,000105 | 0,0102 | 0,000105 |
| 6 | 53400 | 0,178 | 1,288433 | 0,176 | 1,305124 | 1,296778 | 0,0083 | 6,96∙10-5 | 0,0083 | 6,96∙10-5 |
| 7 | 62300 | 0,154 | 1,506015 | 0,151 | 1,536165 | 1,52109 | 0,0150 | 0,000227 | 0,0150 | 0,000227 |
| 8 | 71200 | 0,135 | 1,710406 | 0,133 | 1,733948 | 1,722177 | 0,0117 | 0,000138 | 0,0117 | 0,000138 |
| Сумма: | - | - | - | - | - | - | - | 0,000721 | - | 0,000721 |
Отсюда по формуле (3.15) дисперсия воспроизводимости равна:
![]()
Число степеней свободы ![]() .
.
3.3.3. Проверка адекватности кинетической модели базового опыта.
Проверим модель на адекватность, т.е. ответим на вопрос: можно ли использовать полученное уравнение регрессии или необходима более сложная модель.
а) Для начала нужно найти по формуле (3.16) дисперсию адекватности:
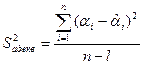 (3.16)
(3.16)
В нашем случае n = 8, а количество значимых коэффициентов уравнения регрессии l
= 1. ![]() найдём по формуле (3.13):
найдём по формуле (3.13): ![]() . Все данные сведены в таблицу (3.5):
. Все данные сведены в таблицу (3.5):
Таблица 3.5. Расчётная таблица для дисперсии адекватности базового опыта.
| i | ||||
| 1 | 0,235566 | 0,217009 | 0,018557 | 0,000344 |
| 2 | 0,419124 | 0,434018 | 0,014894 | 0,000222 |
| 3 | 0,662714 | 0,651027 | 0,011687 | 0,000137 |
| 4 | 0,860113 | 0,868037 | 0,007923 | 6,28∙10-5 |
| 5 | 1,070329 | 1,085046 | 0,014716 | 0,000217 |
| 6 | 1,322055 | 1,302055 | 0,020001 | 0,0004 |
| 7 | 1,515984 | 1,519064 | 0,00308 | 9,49∙10-6 |
| 8 | 1,733948 | 1,736073 | 0,002125 | 4,51∙10-6 |
| Сумма: | - | - | - | 0,001396 |
Тогда по уравнению (3.16):
![]()
Число степеней свободы при этом ![]() .
.
б) Проверку адекватности уравнения регрессии эксперименту проводиться по критерию Фишера по формуле (3.17):
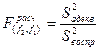 (3.17)
(3.17)
Для нашего случая:
![]()
Для p= 0,05 по табличным данным [1] найдём, что ![]() . Таким образом, т.к.
. Таким образом, т.к. ![]() , то гипотеза об адекватности принимается.
, то гипотеза об адекватности принимается.
3.3.4. Оценка средней квадратичной ошибки коэффициента уравнения регрессии.
По формуле (3.18) имеем:
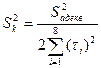 (3.18)
(3.18)
И следовательно:
![]()
3.3.5. Проверка значимости коэффициента уравнения регрессии.
Используя критерий Стьюдента (tj ), проверим значимо ли kотличается от нуля. По формуле (3.19) можно найти расчётный критерий Стьюдента для k:
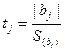 (3.19)
(3.19)
В нашем случае уравнение (3.19) имеет следующий вид:
![]()
Для p = 0,05 и ![]() по таблице квантилей распределения Стьюдента [1] t0,05
(8) = 2,31. А т.к. tj
> t0,05
(8), то нулевая гипотеза отвергается, и следовательно kявляется значимым в уравнении регрессии. Подставив значение kв формулу (3.13), получим линейную кинетическую модель реакции:
по таблице квантилей распределения Стьюдента [1] t0,05
(8) = 2,31. А т.к. tj
> t0,05
(8), то нулевая гипотеза отвергается, и следовательно kявляется значимым в уравнении регрессии. Подставив значение kв формулу (3.13), получим линейную кинетическую модель реакции: ![]() .
.
3.3.6. Нахождение доверительного интервала для k по данному уравнению регрессии для базового опыта.
Найдём доверительный интервал для kпо данному уровню значимости p = 0,05. Для этого используем формулу (3.20):
![]() (3.20)
(3.20)
В нашем случае ![]() для k = 2,41121∙10-5
; t0,05
(8) = 2,31; Sk
= 7,75986∙10-8
. Тогда t0,05
(8)∙Sk
= 2,31∙7,75986∙10-8
= 1,79253∙10-7
. Получили:
для k = 2,41121∙10-5
; t0,05
(8) = 2,31; Sk
= 7,75986∙10-8
. Тогда t0,05
(8)∙Sk
= 2,31∙7,75986∙10-8
= 1,79253∙10-7
. Получили:
![]()
Вывод:
Уравнением ![]() можно пользоваться в пределах эксперимента для описания данной реакции. Поскольку она адекватно описывает опытные данные и хорошо согласуется с экспериментом.
можно пользоваться в пределах эксперимента для описания данной реакции. Поскольку она адекватно описывает опытные данные и хорошо согласуется с экспериментом.
Таким образом, найдена кинетическая модель для описания изучаемой реакции при постоянной температуре. Она имеет вид:
![]()
где D – доверительный интервал константы. Тогда:
![]() .
.
3.3.7. Определение влияния температуры на константу скорости реакции.
Для выяснения влияния температуры на константу скорости реакции, описанным ранее способом находим значение констант скорости при температурах опытов 7, 8, 9:
а) При температуре t=80ºС, СA, O =1,0 моль/л, СY -, O =0,5 моль/л, τ1 = 8900, τ2 = 17800, …, τ8 =71200 были получены такие данные:
Таблица 3.6. Результаты опыта 7, приведённые для линейной функции (3.11).
| i | τi | 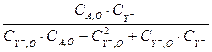 |
αi | |
| 1 | 8900 | 0,911 | 0,902305 | 0,205605 |
| 2 | 17800 | 0,842 | 0,812352 | 0,415644 |
| 3 | 26700 | 0,781 | 0,71959 | 0,658147 |
| 4 | 35600 | 0,736 | 0,641304 | 0,888502 |
| 5 | 44500 | 0,708 | 0,587571 | 1,063518 |
| 6 | 53400 | 0,675 | 0,518519 | 1,313559 |
| 7 | 62300 | 0,651 | 0,463902 | 1,536165 |
| 8 | 71200 | 0,634 | 0,422713 | 1,722124 |
Таблица 3.7. Расчётная таблица для опыта 7.
| i | τi | αi | (τi )2 | τi αi |
| 1 | 8900 | 0,205605 | 79210000 | 1829,885 |
| 2 | 17800 | 0,415644 | 316840000 | 7398,467 |
| 3 | 26700 | 0,658147 | 712890000 | 17572,51 |
| 4 | 35600 | 0,888502 | 1267360000 | 31630,68 |
| 5 | 44500 | 1,063518 | 1980250000 | 47326,54 |
| 6 | 53400 | 1,313559 | 2851560000 | 70144,05 |
| 7 | 62300 | 1,536165 | 3881290000 | 95703,1 |
| 8 | 71200 | 1,722124 | 5069440000 | 122615,2 |
| Сумма: | - | - | 16158840000 | 394220,5 |
Отсюда по формуле (3.14):
![]()
Таблица 3.8. Расчётная таблица для дисперсии адекватности опыта 7.
| i | ||||
| 1 | 0,205605 | 0,21713 | 0,011525 | 0,000133 |
| 2 | 0,415644 | 0,434259 | 0,018615 | 0,000347 |
| 3 | 0,658147 | 0,651389 | 0,006758 | 4,57∙10-5 |
| 4 | 0,888502 | 0,868518 | 0,019984 | 0,000399 |
| 5 | 1,063518 | 1,085648 | 0,02213 | 0,00049 |
| 6 | 1,313559 | 1,302777 | 0,010782 | 0,000116 |
| 7 | 1,536165 | 1,519907 | 0,016258 | 0,000264 |
| 8 | 1,722124 | 1,737037 | 0,014913 | 0,000222 |
| Сумма: | - | - | - | 0,002017 |
Тогда по уравнению (3.16):
![]()
Проверку адекватности уравнения регрессии эксперименту проводиться по критерию Фишера по формуле (3.17):
![]()
Для p= 0,05 по табличным данным [1] , ![]() . Таким образом, т.к.
. Таким образом, т.к. ![]() , то гипотеза об адекватности принимается.
, то гипотеза об адекватности принимается.
б) При температуре t=90ºС, СA, O =1,0 моль/л, СY -, O =0,5 моль/л, τ1 = 3900, τ2 = 7800, …, τ8 =31200 были получены такие данные:
Таблица 3.9. Результаты опыта 8, приведённые для линейной функции (3.11).
| i | τi | |
αi | |
| 1 | 3900 | 0,898 | 0,886414 | 0,241142 |
| 2 | 7800 | 0,841 | 0,810939 | 0,419124 |
| 3 | 11700 | 0,781 | 0,71959 | 0,658147 |
| 4 | 15600 | 0,74 | 0,648649 | 0,865728 |
| 5 | 19500 | 0,702 | 0,575499 | 1,105037 |
| 6 | 23400 | 0,677 | 0,522895 | 1,296749 |
| 7 | 27300 | 0,654 | 0,470948 | 1,506015 |
| 8 | 31200 | 0,633 | 0,420221 | 1,733948 |
Таблица 3.10. Расчётная таблица для опыта 8.
| i | τi | αi | (τi )2 | τi αi |
| 1 | 3900 | 0,241142 | 15210000 | 940,4528832 |
| 2 | 7800 | 0,419124 | 60840000 | 3269,167233 |
| 3 | 11700 | 0,658147 | 137000000 | 7700,315219 |
| 4 | 15600 | 0,865728 | 243000000 | 13505,35937 |
| 5 | 19500 | 1,105037 | 380000000 | 21548,22252 |
| 6 | 23400 | 1,296749 | 548000000 | 30343,92004 |
| 7 | 27300 | 1,506015 | 745000000 | 41114,21324 |
| 8 | 31200 | 1,733948 | 973000000 | 54099,18467 |
| Сумма: | - | - | 3100000000 | 172520,8352 |
Отсюда по формуле (3.14):
![]()
Таблица 3.11. Расчётная таблица для дисперсии адекватности опыта 8.
| i | ||||
| 1 | 0,241142 | 0,216844 | 0,024298 | 0,00059 |
| 2 | 0,419124 | 0,433687 | 0,014563 | 0,000212 |
| 3 | 0,658147 | 0,650531 | 0,007616 | 5,8∙10-5 |
| 4 | 0,865728 | 0,867375 | 0,001647 | 2,71∙10-6 |
| 5 | 1,105037 | 1,084218 | 0,020819 | 0,000433 |
| 6 | 1,296749 | 1,301062 | 0,004313 | 1,86∙10-5 |
| 7 | 1,506015 | 1,517906 | 0,011891 | 0,000141 |
| 8 | 1,733948 | 1,734749 | 0,000801 | 6,42∙10-7 |
| Сумма: | - | - | - | 0,001457 |
Тогда по уравнению (3.16):
![]()
Проверку адекватности уравнения регрессии эксперименту проводиться по критерию Фишера по формуле (3.17):
![]()
Для p= 0,05 по табличным данным [1] , ![]() . Таким образом, т.к.
. Таким образом, т.к. ![]() , то гипотеза об адекватности принимается.
, то гипотеза об адекватности принимается.
в) При температуре t=100ºС, СA, O =1,0 моль/л, СY -, O =0,5 моль/л, τ1 = 1800, τ2 = 3600,…, τ8 =14400 были получены такие данные:
Таблица 3.12. Результаты опыта 9, приведённые для линейной функции (3.11).
| i | τi | |
αi | |
| 1 | 1800 | 0,907 | 0,897464 | 0,216364 |
| 2 | 3600 | 0,833 | 0,79952 | 0,447488 |
| 3 | 5400 | 0,776 | 0,71134 | 0,681209 |
| 4 | 7200 | 0,739 | 0,64682 | 0,871374 |
| 5 | 9000 | 0,703 | 0,577525 | 1,098007 |
| 6 | 10800 | 0,672 | 0,511905 | 1,339233 |
| 7 | 12600 | 0,648 | 0,45679 | 1,567062 |
| 8 | 14400 | 0,63 | 0,412698 | 1,770076 |
Таблица 3.13. Расчётная таблица для опыта 9.
| i | τi | αi | (τi )2 | τi αi |
| 1 | 1800 | 0,216364 | 3240000 | 389,4555 |
| 2 | 3600 | 0,447488 | 12960000 | 1610,957 |
| 3 | 5400 | 0,681209 | 29160000 | 3678,528 |
| 4 | 7200 | 0,871374 | 51840000 | 6273,896 |
| 5 | 9000 | 1,098007 | 81000000 | 9882,067 |
| 6 | 10800 | 1,339233 | 117000000 | 14463,72 |
| 7 | 12600 | 1,567062 | 159000000 | 19744,99 |
| 8 | 14400 | 1,770076 | 207000000 | 25489,1 |
| Сумма: | - | - | 661000000 | 81532,71 |
Отсюда по формуле (3.14):
![]()
Таблица 3.14. Расчётная таблица для дисперсии адекватности опыта 9.
| i | ||||
| 1 | 0,216364 | 0,222039 | 0,005675 | 3,22∙10-5 |
| 2 | 0,447488 | 0,444078 | 0,00341 | 1,16∙10-5 |
| 3 | 0,681209 | 0,666117 | 0,015092 | 0,000228 |
| 4 | 0,871374 | 0,888156 | 0,016782 | 0,000282 |
| 5 | 1,098007 | 1,110195 | 0,012187 | 0,000149 |
| 6 | 1,339233 | 1,332234 | 0,007 | 4,9∙10-5 |
| 7 | 1,567062 | 1,554273 | 0,01279 | 0,000164 |
| 8 | 1,770076 | 1,776312 | 0,006235 | 3,89∙10-5 |
| Сумма: | - | - | - | 0,000953 |
Тогда по уравнению (3.16):
![]()
Проверку адекватности уравнения регрессии эксперименту проводиться по критерию Фишера по формуле (3.17):
![]()
Для p= 0,05 по табличным данным [1] , ![]() . Таким образом, т.к.
. Таким образом, т.к. ![]() , то гипотеза об адекватности принимается.
, то гипотеза об адекватности принимается.
Далее из уравнения Аррениуса (3.21):
![]() (3.21)
(3.21)
Получим линеаризованную форму:
![]() (3.22)
(3.22)
Для найденных значений k7 , k8 , k9 , равных, соответственно:
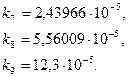
Строим график в координатах уравнения (3.22), т.е. (lnki
) от (1/T). По графику рис. 3.4 можно определить значение Aи значение E, т.к. ![]() , а отрезок отсекаемый графиком на оси lnki
равен lnA. Отсюда, т.к.
, а отрезок отсекаемый графиком на оси lnki
равен lnA. Отсюда, т.к. ![]() , то
, то ![]() , а A = 3,2522∙108
.
, а A = 3,2522∙108
.
По найденному значению энергии активации и значению константы скорости, находим значение энтропии активации, например для опыта 7, по формуле (3.23):
![]() (3.23)
(3.23)
Где NA – число Авогадро, h– постоянная Планка. Следовательно, имеем:
![]()
Полученное положительное значение энтропии активации подтверждает предположение о бимолекулярном механизме реакции.
С учётом полученных активационных параметров кинетическая модель реакции будет иметь вид:
![]() (3.24)
(3.24)
3.4. Проверка адекватности кинетической модели эксперименту.
Проверим модель на адекватность по специальному опыту, который проводился совершенно при другой температуре и концентрациях, т.е. ответим на вопрос: можно ли использовать полученную кинетическую модель или необходима более сложная модель.
а) Для начала нужно найти по формуле (3.16) дисперсию адекватности:
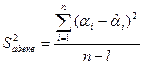 (3.16)
(3.16)
В нашем случае n = 8, а количество значимых коэффициентов уравнения регрессии l
= 1. ![]() найдём по формуле (3.13):
найдём по формуле (3.13): ![]() при следующих условиях: t=110ºС, СA,
O
=0,95 моль/л, СY
-,
O
=0,4 моль/л, τ1
= 840, τ2
= 1680,…, τ8
=6720. Все данные сведены в таблицу (3.15):
при следующих условиях: t=110ºС, СA,
O
=0,95 моль/л, СY
-,
O
=0,4 моль/л, τ1
= 840, τ2
= 1680,…, τ8
=6720. Все данные сведены в таблицу (3.15):
Таблица 3.15. Расчётная таблица для дисперсии адекватности кинетической модели.
| i | τi | ||||
| 1 | 840 | 0,214054 | 0,223192 | 0,009138 | 8,35∙10-5 |
| 2 | 1680 | 0,456142 | 0,446384 | 0,009758 | 9,52∙10-5 |
| 3 | 2520 | 0,670218 | 0,669576 | 0,000642 | 4,12∙10-7 |
| 4 | 3360 | 0,86434 | 0,892768 | 0,028428 | 0,000808 |
| 5 | 4200 | 1,101853 | 1,11596 | 0,014108 | 0,000199 |
| 6 | 5040 | 1,327367 | 1,339152 | 0,011786 | 0,000139 |
| 7 | 5880 | 1,529527 | 1,562345 | 0,032818 | 0,001077 |
| 8 | 6720 | 1,785183 | 1,785537 | 0,000354 | 1,25∙10-7 |
| Сумма: | - | - | - | - | 0,002402 |
Тогда по уравнению (3.16):
![]()
Число степеней свободы при этом ![]() .
.
б) Проверку адекватности уравнения регрессии эксперименту проводиться по критерию Фишера по формуле (3.17):
(3.17)
Для нашего случая:
![]()
Для p= 0,05 по табличным данным [1] найдём, что ![]() . Таким образом, т.к.
. Таким образом, т.к. ![]() , то гипотеза об адекватности принимается.
, то гипотеза об адекватности принимается.
4. Экспериментальная часть
4.1. Выбор метода анализа ключевого компонента.
При диссоциации ключевого компонента образуется два иона:
![]()
Т.е. возможно количественное определение ключевого компонента по одному из этих ионов. При этом следует учесть тот факт, что образующийся побочный продукт реакции, тоже диссоциирует:
![]()
Т.е. в растворе присутствуют катионы калия от обеих солей, поэтому раздельное количественное определение их по катиону невозможно. Остаётся анион йода, а т.к. расходующийся на реакцию йод надёжно связан с метильным радикалом, он не будет присутствовать в растворе и мешать определению непрореагировавшего количества анионов йода, а т.к. диссоциация идёт в мольном соотношении, то следовательно и количества непрореагировавшего ключевого компонента.
Анион йода, при совместном присутствии аниона хлора, наиболее быстро, надёжно и относительно дёшево можно определить осадительным титрованием, а, в частности, аргентометрией [5].
Но для повышения точности анализа вследствие замены недостаточно точных визуальных индикаторов на показания, которые фиксирует прибор, наилучшим методом определения в данном случае будет потенциометрическое осадительное титрование [6].
Теоретические основы метода . Потенциометрический метод анализа основан на измерении потенциала электрода, опущенного в исследуемый раствор, и нахождении зависимости между его величиной и концентрацией определяемого компонента в растворе.
Практически невозможно измерить потенциал одного электрода, поэтому составляют гальванический элемент:
Измерительный | Исследуемый раствор | Электрод сравнения
(индикаторный) электрод | |
И измеряют э.д.с. элемента, то есть разность потенциалов двух электродов, опущенных в исследуемый раствор: э.д.с. = E1 – Е2 . При условия постоянства потенциала электрода сравнения и определенной, заведомо известной величины его, по величине э.д.с. можно определить потенциал индикаторного электрода. Но измеряемое напряжение V k (клеммовое напряжение) будет меньше разницы электродных потенциалов (э.д.с.) на величину падения напряжение IR i :
![]() (4.1)
(4.1)
Где Rj – внутреннее сопротивление ячейки.
Точное измерение э.д.с. возможно лишь в том случае, когда I = 0 , то есть в отсутствии тока. Этого можно добиться встречным включением напряжения, соответствующего э.д.с. (компенсационный метод Поггендорфа), или применением измерительных приборов с высоким входным сопротивлением.
В компенсационном методе на электроды ячейки налагают э.д.с. внешнего источника постоянного тока (батареи или аккумулятора) противоположно направленную э.д.с. гальванической ячейки. При установившейся компенсации, когда э.д.с. ячейки и источника равны, в цепи тока нет. Величину компенсирующей э.д.с. определяют, сравнивая ее с э.д.с. нормального элемента Вестона. На компенсационном методе основан принцип работы потенциометров, например, типа Р - 307 и др. Для измерений в неводных средах, со стеклянными и некоторыми другими электродами, имеющими высокое сопротивление, эти приборы малопригодны.
Другой способ заключается в использовании приборов с большим (до 1011 – 1015 Ом) входным (внешним) сопротивлением, благодаря чему измерения можно проводить практически без мешающего действия тока. Этот способ воплощен в рН–метрах, ионометрах, электронных вольтметрах. Эти приборы удобны в работе, так как измерения проводятся быстро и результаты измерений можно считывать со шкалы (в рН–метрах, ионометрах) или цифрового индикатора (в электронных вольтметрах) [7].
Электроды сравнения . Согласно электрохимическим реакциям, протекающим на поверхности раздела металл-раствор, различают электроды первого, второго и третьего рода.
К электродам первого рода относятся металлы, потенциалы которых обратимы относительно своих ионов, как, например, серебряный электрод в растворе содержащем катионы серебра.
Потенциалы электродов второго рода обратимы относительно своих анионов, образующих с катионами металла электрода малорастворимый осадок; например, серебряный электрод в насыщенном растворе хлорида серебра в присутствии анионов хлора.
Электроды третьего рода представляют собой металлы, которые находятся в равновесии с раствором, насыщенном двумя малорастворимыми электролитами с одним общим анионом и катионами, один из которых является ионом металла электрода, а второй посторонним, находящимся в избытке [6].
По международному соглашению в качестве стандартного электрода принят стандартный водородный электрод, потенциал которого при любой температуре считают равным нулю. Но водородный электрод малопригоден в обычных условиях работы и на практике используются специально изготовленные электроды сравнения, в основном это насыщенный каломельный и хлорсеребряный электроды.
Электрод сравнения должен иметь постоянный, воспроизводимый потенциал даже в условиях слабых токов. Их обычно изготавливают в виде компактных полуэлементов, чтобы можно было погружать непосредственно в анализируемый раствор; роль солевого мостика играет очень небольшое отверстие (точечное отверстие; узкая щель) во внешней оболочке. Потенциалы этих электродов при их изготовлении измеряют относительно стандартного водородного электрода и дают в характеристике (паспорте) электрода.
Насыщенный каломельный и хлорсеребряный электроды применяют как электроды сравнения практически во всех случаях, так как потенциалы их не зависят от состава изучаемого раствора [7].
Индикаторные электроды . Электрод, потенциал которого зависит от концентрации определяемого иона в растворе, называется индикаторным (измерительным).
Зависимость величины электродного потенциала от концентрации ионов в растворе, к которым чувствителен электрод, в идеальном случае выражается уравнением Нернста:
![]() (4.2)
(4.2)
где φ0 - стандартный электродный потенциал; R - газовая постоянная; Т - абсолютная температура; n - заряд иона; F - число Фарадея; а i - активность иона.
В разбавленных растворах коэффициент активности близок к единице, поэтому активность можно заменить концентрацией:
![]() (4.3)
(4.3)
При 25°C и соответствующих значениях R , F , переходя от натуральных логарифмов к десятичным, получим:
![]() (4.4)
(4.4)
Одним из основных требований к индикаторным электродам, кроме определенной зависимости от активности (концентрации), является воспроизводимость и быстрота отклика потенциала при изменении активности иона.
Известны два основных вида индикаторных электродов – металлические и мембранные.
Металлические индикаторные электроды можно изготавливать из различных металлов, способных давать обратимые полуреакции, например, из серебра, меди, ртути, свинца, кадмия. Потенциалы этих металлов воспроизводимо и предсказуемо отражают активность их ионов в растворе. Металлические электроды служат не только для определения собственных ионов; косвенно они чувствительны к анионам, образующим малорастворимые осадки с этими ионами. В этом случае необходимо только насытить изучаемый раствор малорастворимой солью. Например, серебряный электрод, чувствительный к ионам серебра, будет правильно отражать и концентрацию хлорид ионов в растворе, насыщенном хлоридом серебра. И, если по отношению к ионам Ag+ он будет электродом первого рода, то при определении Сl- - электродом второго рода, поскольку он измеряет концентрацию ионов, не участвующих непосредственно в процессе переноса электронов, то есть в электродной реакции.
В окислительно-восстановительных реакциях в качестве индикаторных электродов применяют инертные металлы (платину, золото), потенциал которых зависит от отношения концентраций окисленной и восстановленной форм одного или нескольких веществ в растворе. Металлические электроды изготавливают из проволоки или пластинки. Перед работой поверхность их тщательно очищают. Хорошим способом очистки для многих электродов является быстрое погружение их в концентрированную азотную кислоту и последующее многократное промывание дистиллированной водой.
В отличие от рассмотренных электродов, потенциал которых определяется процессами переноса электронов между ионами в растворе и металлом (то есть электрохимической реакцией), в мембранных ионоселективных электродах возникновение потенциала связано с процессом обмена ионов между мембраной и раствором.
Процесс переноса через мембрану состоит из двух фаз: проникновение иона в мембрану и перемещение иона внутри мембраны. При этом на поверхности возникает потенциал, который начинает препятствовать дальнейшему перемещению ионов и, в конечном счете, устанавливается динамическое равновесие, при котором устанавливается потенциал, подчиняющийся уравнению Нернста. Мембранный электрод – это устройство цилиндрической трубчатой формы, в котором ионоселективная мембрана разделяет внутренний и внешний (анализируемый) растворы и одновременно служит средством электролитического контакта между ними.
Во внутренний раствор постоянного состава, содержащий ионы, к которым селективна мембрана, опущен токоотводящий электрод, обеспечивающий контакт мембранного электрода с измерительным прибором. Причем для токоотводящего электрода (обычно это платиновая или серебряная проволока) созданы условия постоянства его потенциала.
Существующие ионоселективные электроды можно разделить на электроды с твердой мембраной, жидкой мембраной и стеклянные электроды.
Активный компонент твердой мембраны - малорастворимое соединение (в виде моно- или поликристаллов, осадков, твердых ионообменников и др.), обладающие ионной проводимостью. Электроды с такой мембраной "откликающиеся" на одноименные ионы (один из ионов, входящих в состав самого компонента) и часто еще на противоионы связанные с первыми величиной ПР.
Жидкостная мембрана представляет собой не смешивающуюся с водой органическую жидкость (растворитель с растворенным в нем ионообменным веществом), которая обладает селективным свойством проникновения через нее определенных ионов. Органическая и водная фазы в электроде отделены друг от друга полупроницаемой, инертной мембраной. Известны катионо- и анионоселективные жидкие мембраны. Основное отличие жидких мембран от твердых заключается в том, что они содержат подвижные ионогенные группы.
Стеклянные электроды, хотя и имеют твердую мембрану из ионоселективного стекла, по механизму ближе (аналогичны) электродам с жидкой мембраной.
Широкое применение получили рН – чувствительные стеклянные электроды. Кроме того, разработаны различные сорта ионоселективных стекол, способных к обмену с соответствующими катионами (в основном однозарядными), из которых изготавливают катиончувствительные электроды.
Потенциометрические методы анализа можно разделить на прямую потенциометрию (ионометрию) и потенциометрическое титрование.
Прямая потенциометрия . Уравнение Нернста даст простое соотношение между потенциалом электрода и активностью соответствующего иона в растворе. Поэтому на основания измеренной э.д.с. гальванического элемента, зная потенциал электрода сравнения, можно вычислить потенциал индикаторного электрода, а затем рассчитать активность и концентрацию определяемого иона. Однако здесь возникают некоторые трудности.
Во-первых, реально измеряемая э.д.с. гальванического элемента включает в себя кроме потенциалов электродов и диффузионный потенциал (потенциал жидкостного соединения), который возникает между анализируемым раствором и внешним раствором электрода сравнения. Этот потенциал может достигать десятков милливольт, но точно измерить или оценить теоретичёски его невозможно. Его можно свести к минимуму, используя для соединения между растворами солевой мостик, состоящий из концентрированного раствора электролита, ионы которого имеют одинаковую подвижность, например, насыщенного раствора хлорида калия. При этом потенциал жидкостного соединения составляет обычно несколько милливольт или меньше, то есть имеет несущественную для большинства электроаналитических методов величину, за исключением прямой потенциометрии, так как изменение потенциала (или погрешность в измеряемой э.д.с.) даже на 1 мВ дает относительную ошибку до 4 %.
Во-вторых, для вычисления активности определяемого иона по уравнению Нернста надо знать величину стандартного потенциала индикаторного электрода, однако для многих электродов в первую очередь это касается мембранных (которые в основном и используются как индикаторные в ионометрии), это неизвестный потенциал ассиметрии, величина которого изменяется во времени.
В-третьих, из уравнения Нернста можно определить активность, а для вычисления концентрации определяемого вещества требуется знание коэффициентов активности, которые, как правило, недоступны, поскольку обычно состав, а значит и ионная сила раствора, неизвестны. Поэтому при использовании прямой потенциометрии для аналитических целей применяют, обычно, эмпирическую калибровку измерительного электрода.
К достоинствам прямой потенциометрии следует отнести возможность анализа чрезвычайно малых проб и отсутствие изменения или разложения пробы.
Очень ценным качеством прямой потенциометрии (это особенно касается метода калибровки) является возможность автоматизации измерений, а следовательно, и автоматизации контроля производства, уровня загрязнения в производстве, сточных вод и т.д.
Еще более широкое применение в аналитической химии нашло потенциометрическое титрование , которое заключается в потенциометрическом наблюдении за ходом химической реакции. Потенциометрическое титрование можно охарактеризовать как титрование, при котором изменение э.д.с. гальванического элемента записывают в виде функции добавленного титранта.
Для этого необходимо только, чтобы в реакции участвовал ион, для которого существует подходящий индикаторный электрод.
Установка для проведения потенциометрического титрования проста. Она включает в себя сосуд для титрования (химический стакан), в который помещают анализируемый (титруемый) раствор, подходящие электроды – индикаторный и сравнения, опущенные в этот раствор, приспособление для эффективного перемешивания титруемого раствора (обычно, это магнитная мешалка), бюретку со стандартным раствором титранта и измерительный прибор.
Титрант в начале титрования можно добавлять довольно быстро. При приближении к моменту эквивалентности его следует вводить малыми порциями и ждать установления равновесия перед добавлением новой порции, так как необходимо, время для приобретения индикаторным электродом устойчивого потенциала. В противоположность прямой потенциометрии при титровании измерение э.д.с. с высокой точностью не сложно, так как изменение потенциала индикаторного электрода вблизи точки эквивалентности довольно большое.
Главная цель этого метода – установление с высокой точностью точки эквивалентности, то есть объема титранта, точно известной концентрации, пошедшего на взаимодействие с определяемым веществом. Зная эквивалентный объем титранта, рассчитывают количество определяемого вещества по формуле:
![]() (4.5)
(4.5)
где VT - эквивалентный объем титранта, мл; NT - концентрация титранта, н; Э - эквивалент определяемого вещества.
Для определения точки эквивалентности применяют различные инструментальные, графические и расчетные методы. Выбор метода зависит от удобства его применения, характера кривой титрования и допустимой погрешности определения.
Один из простых и удобных методов определения точки эквивалентности, который чаще всего и применяется - нахождение ее по кривой титрования, построенной в координатах: э.д.с. - объем титранта. Причем значения э.д.с. могут быть выражены как в единицах напряжения (мВ, В), так и в других условных единицах рН, деления шкалы - L и др.). Точку эквивалентности определяют, как обычно, на середине скачка титрования. Если скачок не ярко выражен, что наблюдается, например, при титровании разбавленных растворов, то более точно точку эквивалентности находят по дифференциальной кривой титрования, построенной в координатах ∆Е/∆V - V (∆рН/∆V - V, ∆L/∆V - V ), которая в точке эквивалентности дает резкий максимум.
Если э.д.с, соответствующую точке эквивалентности, установить предварительно из подобных титрований или из теоретических расчетов, можно просто титровать до тех пор, пока э.д.с. не приобретет необходимое значение. Этот принцип заложен в основу автоматического потенциометрического титрования.
Потенциометрическое титрование по своим возможностям значительно превосходит титрование с цветными химическими индикаторами, обладая целым рядом преимуществ:
1. более высокой точностью определения, так как субъективная оценка конца титрования заменяется объективными показателями чувствительных приборов;
2. большой чувствительностью, позволяющей определять очень малые количества веществ;
3. возможностью титровать мутные иди окрашенные растворы, исключающие использование индикаторов;
4. возможностью дифференцированно и последовательно определить два и более компонентов в данной порции исследуемого раствора;
5. возможностью автоматизировать титрование, что позволяет уменьшить трудоемкость метода;
6. возможностью получить и термодинамическую информацию о константах диссоциации слабых электролитов, константах образования комплексных ионов.
В потенциометрическом титровании нет необходимости знать потенциал электрода сравнения, стандартные потенциалы индикаторных электродов и значения жидкостных диффузионных потенциалов. В то же время потенциометрическое титрование позволяет:
1. использовать для определения большее число индикаторных электродов, так как во многих случаях требования к электродам в отношении постоянства угла наклона электродной функции и стандартного потенциала менее жесткие, потому что важно не абсолютное значение потенциала (и э.д.с. элемента), а его изменение, связанное с добавленным титрантом;
2. вести измерение в присутствии мешающих веществ, влияющих на потенциал индикаторного электрода, путем подбора титранта, селективно реагирующего только с определяемым веществом.
Широкие возможности объясняются тем, что в потенциометрическом титровании могут быть использованы все четыре типа реакций: кислотно-основные, осаждения, комплексообразования и окисления-восстановления.
Выбор индикаторного электрода при потенциометрическом титровании определяется типом протекающей реакции, либо природой определяемых ионов или ионов титранта, участвующих в реакции, а также удобством работы с электродами.
При окислительно-восстановительном титровании в качестве индикаторных используют электрод из платины или другого благородного металла, при кислотно-основном титровании изменение концентрации ионов водорода измеряют с помощью рН - чувствительных электродов, чаще всего стеклянных, в реакциях осаждения и комплексообразования выбирают электрод, чувствительный к определяемому иону или реагирующему с ним.
Для того чтобы провести анализ потенциометрическим титрованием, необходимо:
1. анализируемый объект (пробу) перевести в раствор;
2. выбрать соответствующий титрант, то есть подобрать химическую реакцию для определяемого вещества, отвечающую требованиям, предъявленным к реакциям в титриметрии;
3. создать условия для проведения данной реакции (рН , t , отсутствие мешающих определению компонентов);
4. подобрать правильно электроды. Так как в качестве электродов сравнения обычно используются каломельный или хлорсеребряный электроды, то задача сводится к правильному выбору индикаторного электрода;
5. перенести анализируемый раствор или его определенную долю (аликвотную часть) в сосуд для титрования и опустить туда электроды, подсоединив их к измерительному прибору;
6. подвести бюретку с титрантом к сосуду для титрования и титровать при эффективном перемешивании, записывая результаты измерений объема добавляемого титранта по бюретке и э.д.с. (рН или L ) по шкале измерительного прибора;
7. по данным измерений построить графическую зависимость э.д.с. (pH, L ) от объема прибавляемого титранта (кривую титрования), определить точку эквивалентности, то есть эквивалентный объем титранта, пошедшего на взаимодействие с определяемым веществом, и рассчитать его количество в граммах по формуле (4.5).
Осадительное потенциометрическое титрование . Осадительное титрование основано на реакции осаждения определяемого вещества (иона) в процессе титрования. При подборе реакции осаждения (титранта) надо руководствоваться требованиями к титриметрическим реакциям и законом, управляющим процессом осаждения, - правилом произведения растворимости. Необходимо создать все условия, чтобы концентрация определяемого (осаждаемого) иона, оставшегося в растворе после осаждения, не превышала 10-6 моль/л.
Основным параметром, позволяющим контролировать ход реакции, является концентрация осаждаемого иона или иона осадителя. Потенциометрический контроль реакции, то есть потенциометрическое титрование возможно, если есть возможность подобрать соответствующий индикаторный электрод.
В осадительном титровании индикаторными электродами могут быть мембранные электроды, чувствительные к одному из ионов, участвующих в реакции осаждения, или металлические электроды, чувствительные к собственному (одноименному) иону, если этот ион осаждается или осаждает: серебряный электрод для определения Ag( I) , ртутный - для определения Hg(II) , медный - для определения Cu(II) и др. Например, серебряный электрод (серебряная проволока или пластина) может служить индикаторным при определении серебра, а также некоторых ионов (С l- , В r- , I- , CN- , CNS- и др.), образующих малорастворимые соли серебра при титровании их раствором азотнокислого серебра.
Электродом сравнения могут служить каломельный и хлорсеребряный электроды. Если в реакции осаждения участвуют ионы хлора, то для предотвращения диффузии хлорида калия в пробу и уменьшения погрешности электрод сравнения можно поместить в отдельный раствор, соединив его с титруемым солевым мостиком из нитрата калия [7].
Определение содержания галогенидов в растворе . Основной задачей является определение хлоридов и иодидов в растворе при совместном присутствии. Для решения ее проводятся два потенциометрических титрования.
1) титрование раствора AgNO3 стандартным раствором NaCl для определения концентрации, то есть для стандартизации раствора AgNO3 ;
2) титрование анализируемого раствора (С l- и I- ) раствором AgNO3 , концентрация которого установлена первым титрованием.
В основе первого титрования лежит реакция:
Ag+ + C l- = AgCl
При титровании смеси иодидов и хлоридов:
I- + Ag+ = AgI
С l- + Ag+ = AgCl
Значительная разница ПР (![]() ) позволяет сделать выводы:
) позволяет сделать выводы:
1) вначале титруется I- с образованием менее растворимого AgI , а затем – С l- ;
2) С
l-
начинает осаждаться только тогда, когда I-
будет полностью осажден (оттитрован), и концентрация Ag+
достигнет величины, необходимой для достижения и превышения ПР
AgCl
: ![]() ;
;
3) получится два четко раздельных скачка на кривой титрования, соответствующие: первый – оттитровыванию I- , второй – С l- (рис. 4.3).
В работе используется серебряный электрод как индикаторный, так как потенциал его определяется концентрацией ионов серебра:
![]() (4.6)
(4.6)
![]() (4.7)
(4.7)
где φ0 Ag = 0,8 В , [ Ag+ ] - концентрация ионов серебра в данный момент титрования, г-ион/л.
При титровании раствора AgNO3 раствором NaCl концентрация ионов серебра уменьшается, и потенциал серебряного электрода будет соответственно уменьшаться. Можно теоретически рассчитать изменение потенциала серебряного электрода (а также э.д.с. гальванического элемента) в процессе титрования, и экспериментально это подтвердить потенциометрическими измерениями.
До прибавления эквивалентного объема NaCl, т.е. до точки эквивалентности потенциал серебряного электрода рассчитывается по уравнению (4.7), где [
Ag+
]
– концентрация ионов серебра, еще не вступивших в реакцию с хлоридом; в момент эквивалентности, при прибавлении эквивалентного количества осадителя ![]() и поэтому
и поэтому
![]() (4.8)
(4.8)
после точки эквивалентности при избытке осадителя ![]() , где [
Cl-
]
определяется избытком титранта
, где [
Cl-
]
определяется избытком титранта
![]() (4.9)
(4.9)
ПР - const, поэтому
![]() (4.10)
(4.10)
Кривая титрования будет иметь вид, изображенный на рис. 4.1.
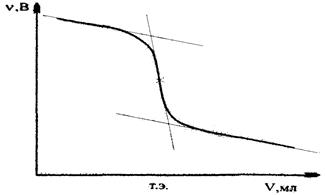
Рис. 4.1. Кривая титрования AgNO3 раствором NaCl .
При титровании галогенида, например С
l-
, раствором AgNO3
потенциал серебряного электрода будет увеличиваться, т.к. концентрация ионов серебра возрастает. До точки эквивалентности, когда переведены в осадок еще не все ионы хлора ![]() , где [
Cl-
]
- концентрация непрореагировавших ионов, г-ион/л.
, где [
Cl-
]
- концентрация непрореагировавших ионов, г-ион/л.
Соответственно потенциал индикаторного электрода находят по уравнению (4.10). В точке эквивалентности, когда ионы хлора практически будут осаждены, а концентрация ионов серебра станет равной концентрации ионов хлора в растворе ![]() , потенциал индикаторного электрода по уравнению (4.8). После точки эквивалентности, концентрация ионов серебра будет определяться избытком титранта и φ
Ag
рассчитывают по (4.7). Кривая титрования показана на рис. 4.2.
, потенциал индикаторного электрода по уравнению (4.8). После точки эквивалентности, концентрация ионов серебра будет определяться избытком титранта и φ
Ag
рассчитывают по (4.7). Кривая титрования показана на рис. 4.2.
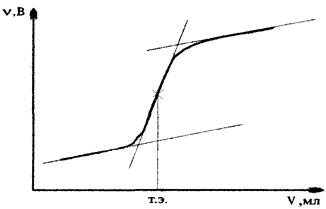
Рис. 4.2. Кривая титрования хлоридов раствором AgNO3 .
При потенциометрическом титровании смеси иодидов и хлоридов получится кривая титрования (рис. 4.3) с двумя скачками, соответствующими моментам эквивалентности реакций осаждения иодидов и хлоридов [7].
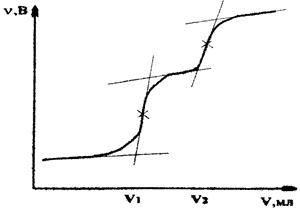
Рис 4.3. Кривая титрования иодидов и хлоридов раствором AgNO3 .
Методика проведения анализа . Дифференцированное определение I- и Cl- в их смеси проводят титрованием ~ 0,05 н. стандартным раствором нитрата серебра с серебряным индикаторным электродом и Нас.КЭ (насыщенным каломельным электродом) сравнения. Э.д.с. потенциометрической ячейки измеряют компенсационным методом.
Пока в растворе присутствуют ионы иодида, потенциал Нас.КЭ больше потенциала серебряного электрода, поэтому последний должен быть подключен к отрицательной клемме потенциометра [6].
До начала работы необходимо:
1) подключить измерительный прибор к электросети для прогревания в течение 15-20 минут;
2) зачистить наждачной бумагой металлическую часть индикаторного (серебряного) электрода и промыть дистиллированной водой весь электрод;
3) протереть фильтровальной бумагой и промыть дистиллированной водой электрод сравнения;
4) тщательно протереть фильтровальной бумагой и промыть дистиллированной водой магнитный элемент.
Работа состоит из двух частей:
а) Определение методом потенциометрического титрования концентрации раствора
AgN
O3
(![]() ).
).
В чистый стакан для титрования отобрать с помощью бюретки 3 мл раствора AgN O3 поместить в стакан магнитный элемент и опустить электроды так, чтобы они не касались магнитного элемента в покое и в дальнейшем при перемешивании раствора. Прилить в стакан дистиллированной воды столько, чтобы рабочие части электродов (металлическая часть индикаторного и место контакта с раствором электрода сравнения) были полностью погружены в раствор. Включить магнитную мешалку и подобрать подходящий режим перемешивания. Включить измерительный прибор и измерить начальную величину э.д.с. полученного гальванического элемента. Затем титровать стандартным раствором NaCl , прибавляя его по 0,2 мл и измеряя э.д.с. после каждой порции прибавленного титранта. Полученные измерения заносят в таблицу. В начале титрования э.д.с. мало изменяется, затем вблизи точки, эквивалентности происходит резкое, скачкообразное изменение, после конца скачка опять наблюдаются близкие по значению величины э.д.с. Титровать необходимо до получения 6 - 7 измерений близких по значению величии э.д.с. после конца скачка титрования (~ до 140 - 150 мВ, т.е. 0,14 -0,15 В).
б) Определение методом потенциометрического титрования содержания хлоридов и иодидов при совместном присутствии.
Полученный у лаборанта анализируемый раствор, содержащий смесь солей K I и NaCl , титруют раствором AgN O3 по выше описанной методике. Измерения записывают в таблицу. В отличие от предыдущего титрования здесь должно получиться два скачка титрования, то есть два резких скачкообразных изменения э.д.с. Титрование заканчивают, добавив 6 - 7 порций титранта после конца второго скачка титрования (~ до 420 - 450 мВ, т.е. до 0,42-0,45 В).
По полученным данным строят кривые титрования - графические зависимости э.д.с. от объема (V) титранта (рис. 4.1, 4.2, 4.3). На скачках титрования находят точки эквивалентности и соответствующие им эквивалентные объемы титрантов, израсходованные на взаимодействие с титруемыми веществами.
Рассчитывают:
Концентрацию AgN O3 по формуле:
![]() (4.11)
(4.11)
где ![]() - концентрация стандартного раствора NaCl
, взятого для титрования, н;
- концентрация стандартного раствора NaCl
, взятого для титрования, н; ![]() - объем стандартного раствора NaCl
, пошедший на титрование AgNO3
, мл. (Рис. 4.1);
- объем стандартного раствора NaCl
, пошедший на титрование AgNO3
, мл. (Рис. 4.1);
![]() - объем раствора AgNO3
, отобранный для титрования, 3,00 мл.
- объем раствора AgNO3
, отобранный для титрования, 3,00 мл.
Содержание хлор-иона и иона йода в анализируемом растворе рассчитывают по формулам:
![]() (4.12)
(4.12)
![]() (4.13)
(4.13)
где V1
– объем AgNO3
пошедший на титрование иодида, соответствующий первому скачку на кривой титрования смеси солей, мл (рис 4.3); V2
– общий объем AgN
O3
, пошедший на титрование йодида и хлорида, соответствующий второму скачку на кривой титрования смеси солей, мл (рис. 4.3); ![]() - концентрация раствора AgN
O3
, рассчитанная по формуле (4.11); Э
Cl
-, Э
I
-
- эквиваленты хлора и йода, равные их атомным массам.
- концентрация раствора AgN
O3
, рассчитанная по формуле (4.11); Э
Cl
-, Э
I
-
- эквиваленты хлора и йода, равные их атомным массам.
Содержание KI и KCl а анализируемом растворе рассчитывается но формулам:
![]() (4.14)
(4.14)
![]() (4.15)
(4.15)
где Э KI и Э KCl - эквиваленты KI и KCl , равные их молекулярным массам [7].
4.2. Приготовление растворов реагирующих веществ.
Вещество иодид калия в условиях реакции является кристаллическим хорошо растворимым веществом, поэтому приготовление его раствора заключается в растворении его в растворителе (дистиллировано воде).
Вещество хлористый метан в условиях реакции является газом хорошо растворимым в спирте (3500мл на 100г (127мл) этанола), поэтому приготовление его раствора заключается в растворении его в этаноле или нахождении готового реактива (этанол который содержит уловленный хлористый метанол, концентрация принимается максимально возможной).
Вещество йодистый метан в условиях реакции является жидкостью, бесконечно растворимой в этаноле, поэтому приготовление его раствора заключается в растворении его в растворителе (этаноле).
Объём реакционной массы составляет 720 мл.
Опыт 1 . Т.е. Vр.м. = 720мл, С A,0 = 1,0 моль/л, С Y,0 = 0,5 моль/л, С Z,0 = 0,0 моль/л, tº = 80º C, растворитель – водный раствор спирта.
Для приготовления раствора хлористого метана необходимо взять 585,21 мл готового этанола (с максимальной концентрацией) с растворённым в нём веществом.
![]()
Для приготовления раствора иодистого калия к концентрацией 0,5 моль/л необходимо взвесить 59,76г чистого вещества. Количественно перенести в колбу, долить 134,79 мл дистиллированной воды.
![]()
Опыт 4 . Т.е. Vр.м. = 720мл, С A,0 = 1,0 моль/л, С Y,0 = 0,5 моль/л, С Z,0 = 0,2 моль/л, tº = 80º C, растворитель – водный раствор спирта.
Для приготовления раствора хлористого метана необходимо взять 585,21 мл готового этанола (с максимальной концентрацией) с растворённым в нём веществом.
![]()
Для приготовления раствора иодистого калия к концентрацией 0,5 моль/л необходимо взвесить 59,76г чистого вещества. Количественно перенести в колбу, долить 104,79 мл дистиллированной воды.
![]()
Для приготовления раствора йодистого метана с концентрацией 0,2 моль/л необходимо взять 28,4мл чистого вещества. Количественно перенести его в колбу, долить 30мл чистого этанола.
![]()
4.3. Схема установки, подбор реактора и доказательство его идеальности.
Для проведения кинетических опытов необходим реактор идеального смешения (РИС). Это тип идеального реактора можно получить на следующей установке (пример):
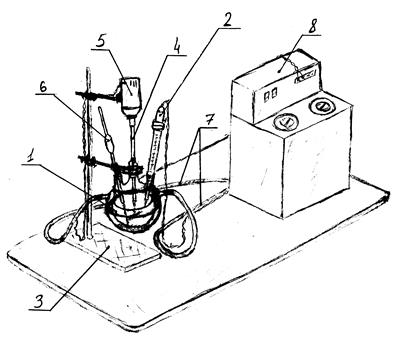
Рис. 4.4. Схема экспериментальной установки.
1 – стеклянный трёхгорлый реактор с рубашкой; 2 – ртутный термометр с контактами (РТК); 3 – штатив; 4 – стеклянная мешалка; 5 – электропривод мешалки; 6 – пипетка для отбора аликвот; 7 – трубки силиконовые; 8 – ультратермостат.
Реактор представляет стеклянный трёхгорлый реактор 1 с напаянной стеклянной рубашкой. В реакторе через центральный шлиф помещена мешалка 4, приводимая в движение электроприводом 5.
Реактор снабжён следующими узлами:
а) узел термостатирования представлен рубашкой реактора, трубками 7, соединяющими её с ультратермостатом 8, который поддерживает постоянную температуру теплоносителя (вода) и прокачивает его через рубашку со скорость, обеспечивающей постоянство температуры в ректоре. Объём рубашки должен составлять не менее ½ объёма реактора. Также дополнительно имеется ртутный термометр с контактами (РТК), посредством которого отслеживается температура реакционной массы, и в случае превышения температуры ультратермостат должен автоматически увеличивать скоростью подачи теплоносителя в рубашку реактора.
б) узел моментального ввода реагентов представляет собой мерный цилиндр и стеклянную воронку, посредством которой через свободный шлиф в реактор вводиться второй реагент. Такая реализация этого узла возможна, поскольку константа скорости протекания реакции очень мала и реакция протекает медленно. Т.е. время ввода второго реагента по сравнению со временем протекания реакции совершенно незначительно, т.е. практически мгновенно.
в) узел моментального отбора проб представляет собой пипетку с резиновой грушей и химический стаканчик для взятой аликвоты. Аналогично предыдущему пункту можно считать время отбора пробы совершенно незначительным.
Учитывая дальнейшее прохождение реакции во взятой аликвоте наиболее удобным и надёжным методом стопорения реакции является метод захолаживания. Т.е. так как скорость протекания реакции мала, и она очень сильно зависит от температуры, то даже при охлаждении аликвоты базового опыта проточной водой падение температуры составляет ~ 60ºC. При таком падении температуры реакция практически перестаёт идти. Но для получения более высокой точности возможно использование смеси воды со льдом, для последующего охлаждения аликвоты.
Подбор объёма реактора . Поскольку для определения кинетики данной реакции наиболее подходит РИС, то необходимо определить его объём.
В плане экспериментов принято для построения кривой 8 точек, для каждой из которых по требованиям аналитической химии выполняется 3 раза. Следовательно, т.к. анализ требует аликвоту объёмом 3 мл, то для определения одной точки на кривой необходимо отобрать 3 мл x 3 = 9 мл реакционной массы. Отсюда, для проведения полного плана экспериментов необходимо 9 мл x 8 = 72 мл реакционной массы.
Отсюда в соответствии с кинетическим правилом, которое говорит о том, что объём аликвотной части не должен превышать 10% от общего объёма, имеем, что 72 мл / 0,1 = 720 мл. Также учитывая коэффициент заполнения для не пенящихся веществ: 720 мл / 0,8 = 900 мл.
А т.к. наиболее близкий стандартный объём реактора – 1 л., то соответственно для проведения кинетических экспериментов необходим стеклянный реактор объёмом 1л.
Доказательство идеальности реактора.
а) Так как в реакторе организовано эффективное перемешивание и отсутствие застойных зон, вследствие сферической формы реактора, то обеспечивается моментальное распределение второго реагента по объёму реактора. Что доказано при тестировании химическими и визуальными трассерами. При тестировании химическим трассером было показано, что раствор в реакторе приобретает однородную концентрацию по всему объёму в течение малого промежутка времени. А при внесении визуального раствора аналогично раствор приобретал равномерную окраску за очень малый промежуток. Что удовлетворяет условию идеальности проведения данной реакции.
б) Эффективная система термостатирования и эффективная система перемешивания обеспечивает моментальное распределение колебаний температуры по объёму, возможных при подаче второго реагента.
Что было доказано внесением трассеров в реактор с пониженными и повышенными температурами, в результате чего происходило быстрое выравнивание температуры реакционной среды до температуры термостатирования.
в) Организован моментальный ввод второго реагента.
4.4. Прописи кинетических экспериментов.
Опыт 1 .
В реактор, снабжённый обратным холодильником, термометром и мешалкой, помещают приготовленный раствор иодида калия и термостатируют до температуры реакции. Одновременно в термостате термостатируется раствор хлористого метана, который затем приливают стеклянным стаканом через воронку в реактор, момент внесения считается началом реакции. В течении 20 часов, через каждые 150 мин отбирают пробу реакционной массы. Для каждой пробы проводят захолаживание, потенциометрическое титрование и определяют концентрацию ключевого вещества.
С A,0 = 1,0 моль/л, С Y,0 = 0,5 моль/л, С Z,0 = 0,0 моль/л, tº = 80º C.
Номер точки Время Концентрация вещества Y, моль/л
1 9000.0 0.400
2 18000 0.341
3 27000 0.280
4 36000 0.241
5 45000 0.207
6 54000 0.174
7 63000 0.153
8 72000 0.133
Опыт 2 .
С A,0 = 9,0 моль/л, С Y,0 = 0,5 моль/л, С Z,0 = 0,0 моль/л, tº = 80º C.
Номер точки Время Концентрация вещества Y, моль/л
1 9900.0 0.408
2 19800 0.342
3 29700 0.280
4 39600 0.246
5 49500 0.213
6 59400 0.178
7 69300 0.157
8 79200 0.137
Опыт 3 .
С A,0 = 1,0 моль/л, С Y,0 = 0,4 моль/л, С Z,0 = 0,0 моль/л, tº = 80º C.
Номер точки Время Концентрация вещества Y, моль/л
1 8400.0 0.335
2 16800 0.274
3 25200 0.226
4 33600 0.192
5 42000 0.165
6 50400 0.143
7 58800 0.122
8 67200 0.104
Опыт 4 .
В реактор, снабжённый обратным холодильником, термометром и мешалкой, помещают приготовленный раствор иодида калия и термостатируют до температуры реакции. Одновременно в термостате термостатируется раствор хлористого метана и йодистого метана, который затем приливают стеклянным стаканом через воронку в реактор, после него в реактор приливают раствор хлористого метана, момент внесения считается началом реакции. В течении 19,78 часов (1187 мин), через каждые 148,3 мин отбирают пробу реакционной массы. Для каждой пробы проводят захолаживание, потенциометрическое титрование и определяют концентрацию ключевого вещества.
С A,0 = 1,0 моль/л, С Y,0 = 0,5 моль/л, С Z,0 = 0,2 моль/л, tº = 80º C.
Номер точки Время Концентрация вещества Y, моль/л
1 8900.0 0.409
2 17800 0.343
3 26700 0.288
4 35600 0.244
5 44500 0.209
6 53400 0.174
7 62300 0.152
8 71200 0.132
Опыт 5 .
С A,0 = 1,0 моль/л, С Y,0 = 0,5 моль/л, С Z,0 = 0,0 моль/л, tº = 80º C.
Номер точки Время Концентрация вещества Y, моль/л
1 8900.0 0.406
2 17800 0.336
3 26700 0.281
4 35600 0.242
5 44500 0.205
6 53400 0.178
7 62300 0.154
8 71200 0.135
Опыт 6 .
С A,0 = 1,0 моль/л, С Y,0 = 0,5 моль/л, С Z,0 = 0,0 моль/л, tº = 80º C.
Номер точки Время Концентрация вещества Y, моль/л
1 8900.0 0.403
2 17800 0.343
3 26700 0.282
4 35600 0.243
5 44500 0.208
6 53400 0.176
7 62300 0.151
8 71200 0.133
Опыт 7 .
С A,0 = 1,0 моль/л, С Y,0 = 0,5 моль/л, С Z,0 = 0,0 моль/л, tº = 80º C.
Номер точки Время Концентрация вещества Y, моль/л
1 8900.0 0.411
2 17800 0.342
3 26700 0.281
4 35600 0.236
5 44500 0.208
6 53400 0.175
7 62300 0.151
8 71200 0.134
Опыт 8 .
С A,0 = 1,0 моль/л, С Y,0 = 0,5 моль/л, С Z,0 = 0,0 моль/л, tº = 90º C.
Номер точки Время Концентрация вещества Y, моль/л
1 3900.0 0.398
2 7800.0 0.341
3 11700 0.281
4 15600 0.240
5 19500 0.202
6 23400 0.177
7 27300 0.154
8 31200 0.133
Опыт 9 .
С A,0 = 1,0 моль/л, С Y,0 = 0,5 моль/л, С Z,0 = 0,0 моль/л, tº = 100º C.
Номер точки Время Концентрация вещества Y, моль/л
1 1800.0 0.407
2 3600.0 0.333
3 5400.0 0.276
4 7200.0 0.239
5 9000.0 0.203
6 10800 0.172
7 12600 0.148
8 14400 0.130
Опыт 10 .
С A,0 = 0,95 моль/л, С Y,0 = 0,4 моль/л, С Z,0 = 0,125 моль/л, tº = 110º C.
Номер точки Время Концентрация вещества Y, моль/л
1 840.00 0.329
2 1680.0 0.268
3 2520.0 0.226
4 3360.0 0.195
5 4200.0 0.164
6 5040.0 0.140
7 5880.0 0.122
8 6720.0 0.103
Выводы
В результате проведённой работы была найдена кинетическая модель процесса бимолекулярного замещения в реакции хлористого метила и йодида калия: ![]() .
.
При этом значения энергии активации, частотного фактора Аррениуса и энтропии активации имеют следующие значения: A = 3,2522∙108
, ![]() ,
, ![]() .
.
Анализ полученных данных подтвердил гипотезу о протекании реакции по механизму бимолекулярного механизма. Что подтверждается тем, что при использовании полученных математических моделей получено сходимое с эмпирическими значение энтропии активации комплекса.
Список используемой литературы.
1. Ахназарова, С. Л. Методы оптимизации эксперимента в химической технологии: учеб. пособие для хим.-технол. спец. вузов/ С. Л. Ахназарова, В. В. Кафаров. – 2-е изд., перераб. и доп. – М.: Высшая школа, 1985. – 327с.
2. Лебедев, Н. Н. Химия и технология основного органического и нефтехимического синтеза: учебник для вузов/ 4-е изд., перераб. и доп. – М.: Химия, 1988. – 592с.
3. Попов, Ю. В. Инженерная химия: учебное пособие/ Ю. В. Попов, Б. И. Но. – Волгоград: Волгоград. гос. техн. ун.-т, 2003. – 208с.
4. Сайкс, П. Механизмы реакции в органической химии/ Пер. с англ. под ред. проф. Варшавского Я. М. – изд. 3-е. – М.: Химия, 1977. – 320с.
5. Крешков, А.П. Основы аналитической химии. Теоретические основы. Количественный анализ/ А.П. Крешков. – К.2, 3-е изд., пераб. – М.: Химия, 1970. – 456с.
6. Крешков, А.П. Основы аналитической химии. Физико-химические (инструментальные) методы анализа/ А.П. Крешков. – К.3. – М.: Химия, 1970. – 472с.
7. Кокшарова, И.У. Электрохимические методы анализа: учебное пособие/ И.У. Кокшарова. – Волгоград: ВолгГТУ, 2003. – 55с.