| Скачать .docx | Скачать .pdf |
Реферат: Реальные газы
Реферат подготовила Магарамова Инесса
1.Реальные газы
Модель идеального газа, используемая в молекулярно-кинетической теории газов, позволяющая описывать поведение разрежённых реальных газов при достаточно высоких температурах и низких давлениях. При выводе уравнения состояния идеального газа размерами молекул и их взаимодействием друг с другом пренебрегают. Повышение давления приводит к уменьшению среднего расстояния между молекулами, поэтому необходимо учитывать объём молекул и взаимодействие между ними. При высоких давлениях и низких температурах указанная модель идеального газа непригодна.
При рассмотрении реальных газов – газов, свойства которых зависят от взаимодействия молекул, надо учитывать силы межмолекулярного взаимодействия. Они проявляются на расстояниях ≤10-9 м. и быстро убывают при увеличении расстояния между молекулами. Такие силы называются короткодействующими.
В ХХ в., по мере развития и представлений о строении атома и квантовой механики, было выяснено, что между молекулами вещества одновременно действуют силы притяжения и силы отталкивания. Силы отталкивания считаются положительными, а силы взаимного притяжения – отрицательными.
2. Внутренняя энергия реального газа
Внутренняя энергия реального газа складывается из кинетической энергии теплового движения его молекул и из потенциальной энергии межмолекулярного взаимодействия. Потенциальная энергия реального газа обусловлена только силами притяжения между молекулами. Наличие сил притяжения приводит к возникновению внутреннего давления на газ.
р΄=а/V2
Работа, которая затрачивается для преодоления сил притяжения, действующих между молекулами газа, или, иными словами, против внутреннего давления, как известно из механики, идёт на увеличение потенциальной энергии системы.
Т.е. dA=p΄Vm=dП, или dП=a/V2m*dVm, откуда П=-а/Vm.
Знак минус означает, что молекулярные силы, создающие внутреннее давление р΄, являются силами притяжения. Учитывая оба слагаемых, получим, что внутренняя энергия моля реального газа Um=CVT-a/Vm растёт с повышением температуры и увеличением объёма.
Если газ расширяется без теплообмена с окружающей средой и не совершает внешней работы, то на основании первого начала термодинамики получим, что U1=U2. Следовательно, при адиабатическом расширении без совершения внешней работы внутренняя энергия газа не изменяется.
3. Уравнение Ван-дер-Ваальса
Учёт собственного объёма молекул и сил межмолекулярного взаимодействия привёл голландского физика И. Ван-дер-Ваальса (1837-1923) к выводу уравнения состояния реального газа. Ван-дер-Ваальсом в уравнение Клапейрона-Менделеева введены две поправки.
1. Учёт собственного объёма молекул. Наличие сил отталкивания, которые противодействуют проникновению в занятый молекулой объём других молекул, сводится к тому, что фактический свободный объём, в котором могут двигаться молекулы реального газа, будет не Vm, а Vm-b, где b- объём, занимаемый самими молекулами. Объём b равен утверждённому собственному объёму молекул. Если, например, в сосуде находятся две молекулы, то центр любой из них не может приблизиться к центру другой молекулы на расстояние меньше d, это означает, что для центров обеих молекул оказывается недоступным объём сферы радиусом d, объём, равный восьми объёмам молекулы, а в расчёте на одну молекулу – учетверённый объём молекулы.
2. Учёт притяжения молекул. Действие сил притяжения между молекулами реального газа приводит к появлению дополнительного давления на газ, называемого внутренним давлением. По вычислениям Ван-дер-Ваальса, внутреннее давление обратно квадрату объёма газа, т.е. p΄=a/V2 , где а – постоянная Ван-дер-Ваальса, характеризующая силы межмолекулярного притяжения, Vm – молярный объём. Вводя эти поправки – получим уравнение Ван-дер-Ваальса для моля газа (уравнение состояния идеальных газов): (p+a/V2m) (Vm-b)=RT. При выводе уравнения Ван-дер-Ваальса сделан целый ряд упрощений, поэтому оно также весьма приближённое, хотя и лучше согласуется с опытом, чем уравнение состояния идеального газа. При малых давлениях и высоких температурах объём Vm становится большим, поэтому b<<Vm, и уравнение Ван-дер-Ваальса в данном случае совпадает с уравнением Клапейрона-Менделеева.
4. Изотермы Ван-дер-Ваальса
Для исследования поведения реального газа рассмотрим изотермы Ван-дер-Ваальса – кривые зависимости p от Vm при заданных Т, - определяемые уравнением Ван-дер-Ваальса для моля газа. Эти кривые, полученные для четырёх различных температур имеют довольно своеобразный характер: при высоких температурах (Т>Тк) изотерма реального газа отличается от изотермы идеального газа только некоторым искажением её формы, оставаясь монотонно спадающей кривой; при некоторой температуре, на изотерме имеется лишь одна точка перегиба; при низких температурах (Т<Тк) изотермы имеют волнообразный участок, сначала монотонно опускаясь вниз, затем монотонно поднимаясь вверх и снова монотонно опускаясь.
Для пояснения характера изотерм реального газа преобразуем уравнение Ван-дер-Ваальса к виду: pV3m-(RT+pb)V2m+aVm-ab=0. Это уравнение при заданных р и Т Является уравнением третьей степени относительно Vm; следовательно, оно может иметь либо три вещественных корня, либо один вещественный и два мнимых, причём физический смысл имеют лишь вещественные положительные корни. Поэтому первому случаю соответствуют изотермы при низких температурах, второму случаю – изотермы при высоких температурах.
5. Фазовые переходы первого и второго рода
Фазой называется термодинамически равновесное состояние вещества, отличающееся от других возможных равновесных состояний того же вещества. Если, например, в закрытом сосуде находится вода, то эта система является двухфазной: жидкая фаза – вода и газообразная фаза – смесь воздуха с водяными парами. Если в воду бросить кусочки льда, то эта система станет трёхфазной, в которой лёд является твёрдой фазой.
Часто понятие «фаза» употребляется в смысле агрегатного состояния, однако надо учитывать, что оно шире, чем «агрегатное состояние». В пределах одного агрегатного состояния вещество может находиться в нескольких фазах, отличающихся по своим веществам, составу и строению.
Переход вещества от одной фазы в другую – фазовый переход – всегда связан с качественными изменениями свойств вещества. Примером фазового перехода могут служить изменения агрегатного состояния вещества или переходы, связанные с изменениями в составе, строении и свойствах вещества (например, переход кристаллического вещества из одной модификации в другую).
Различают фазовые переходы двух родов. Фазовый переход первого рода (например, плавление, кристаллизация и т.д.) сопровождается поглощением или выделением вполне определённого количества теплоты, называемой теплотой фазового перехода.
Фазовые переходы первого рода характеризуются постоянством температуры, изменениями энтропии и объёма. Объяснение этому можно дать следующим образом. Например, при плавлении телу нужно сообщить некоторое количество теплоты, чтобы вызвать разрушение кристаллической решётки. Подводимая при плавлении теплота идёт не на нагрев тела, а на разрыв межатомных связей, поэтому плавление протекает при постоянной температуре. При подобных переходах – из более упорядоченного кристаллического состояния в менее упорядоченное жидкое состояние – степень беспорядка увеличивается и, с точки зрения второго начала термодинамики, этот процесс связан с возрастанием энтропии системы. Если переход происходит в обратном направлении (кристаллизация), то система теплоту выделяет. В качестве примера на рисунке 1 показана температурная зависимость свободной энергии F, приходящейся на одну молекулу кристалла, при его превращении в пар. Верхняя ветвь отвечает кристаллическому состоянию, а нижняя ветвь представляет свободную энергию парообразной фазы. При низких температурах свободная энергия кристалла меньше, чем пара, и, следовательно, кристаллическое состояние выгоднее. При высоких температурах, наоборот, выгоднее существование парообразного состояния. Штриховыми линиями показаны области метастабильных, термодинамически неустойчивых состояний системы.
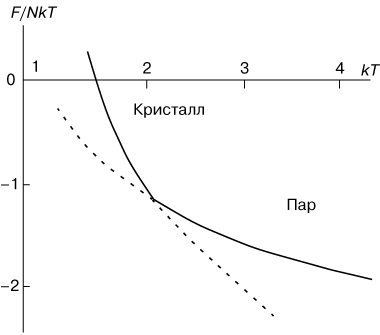 |
| Рис. 1. - Температурная зависимость свободной энергии F при фазовом переходе первого рода "пар-кристалл". |
Поведение внутренней энергии системы, приходящейся на одну молекулу, изображено на рисунке 2. Нижняя ветвь относится к кристаллическому состоянию, а верхняя к парообразному. Скачок энергии в точке перехода представляет собой поглощаемую скрытую теплоту. Соответственно теплоемкость в точке фазового перехода первого рода имеет "всплеск".
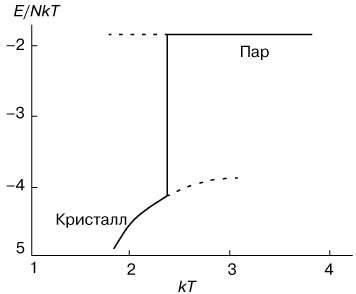 |
| Рис. 2. - Изменение энергии E в зависимости от температуры T при фазовом переходе первого рода "пар-кристалл". |
При теоретическом описании фазовых переходов первого рода каждую из фаз обычно описывают отдельно. Так, кристаллическую ветвь рассматривают, пользуясь моделью идеального кристалла, т. е. предполагая регулярное расположение всех атомов. Парообразную же ветвь получают, используя модель идеального газа, предполагающую полный беспорядок в системе. Зависимости, полученные для различных моделей, накладывают друг на друга и исследуют, какая из возможностей реализуется в данных условиях. Получить описание фазового перехода первого рода, одновременно учитывая все состояния системы, до настоящего времени не удается из-за огромных математических трудностей.
Фазовые переходы, не связанные с поглощением или выделением теплоты и изменением объёма, называются фазовыми переходами второго рода. Эти переходы характеризуются постоянством объёма и энтропии, но скачкообразным изменением теплоёмкости. Общая трактовка фазовых переходов второго рода предложена советским учёным Л.Д.Ландау (1908-1968). Согласно этой трактовке, фазовые переходы второго рода связаны с изменением симметрии: выше точки перехода система, как правило, обладает более высокой симметрией, чем ниже точки перехода. Примерами фазовых переходов второго рода являются: переход ферромагнитных веществ (железа, никеля) при определённых давлении и температуре в парамагнитное состояние; переход металлов и некоторых сплавов при температуре, близкой к 0К, в сверхпроводящее состояние, характеризуемое скачкообразным уменьшением электрического сопротивления до нуля; превращение обыкновенного жидкого гелия при Т=2,9К в другую жидкую модификацию, обладающую свойствами сверхтекучести.
6. Третье начало термодинамики (Теорема Нернста)
Третье начало термодинамики было сформулировано в 1906 году немецким физиком и химиком Вольтером Фридрихом Германом Нернстом (1864 - 1941) эмпирическим путем на основе обобщения экспериментальных данных и получило название теоремы Нернста:
При стремлении температуры любой равновесной термодинамической системы к абсолютному нулю ее энтропия стремится к некоторой универсальной постоянной величине, значение которой не зависит от каких-либо термодинамических параметров системы и может быть принято равной нулю:
| (1) |
Из утверждения теоремы Нернста о независимости значения энтропии равновесной системы при абсолютном нуле температуры от ее термодинамических параметров следует также выражение:
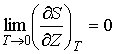 , , |
(2) |
где ![]() - любой термодинамический параметр системы, например, объем, давление и т.д. Здесь нижний индекс
- любой термодинамический параметр системы, например, объем, давление и т.д. Здесь нижний индекс ![]() за скобками обозначает дифференцирование при постоянном значение величины
за скобками обозначает дифференцирование при постоянном значение величины ![]() .
.
Теорема Нернста применима только для систем, находящихся в состоянии термодинамического равновесия и не справедлива для неравновесных систем. В частности, при стремлении температуры аморфного тела, например, стекла, к абсолютному нулю, его энтропия не стремится к некоторому определенному постоянному значению. В зависимости от того, как осуществляется процесс охлаждения, энтропия аморфного тела при стремлении к абсолютному нулю будет различной. Это связано с тем, что для аморфных тел, которые находятся в неравновесном (метастабильном) состоянии, процесс охлаждения может происходить быстрее, чем переход их в равновесное (кристаллическое) состояние.
Из третьего начала термодинамики непосредственно следует недостижимость температуры равной абсолютному нулю. Действительно, для того, чтобы практически осуществить охлаждение термодинамической системы до абсолютного нуля температуры, необходимо чередовать изотермическое сжатие и адиабатическое расширение. При первом процессе происходит отвод теплоты, а при втором - уменьшение температуры системы энтропии.
Другим следствием третьего начала термодинамики является невозможность использования уравнения Клапейрона-Менделеева для описания идеального газа при температурах, близких к абсолютному нулю. Так как для идеального газа на основании первого начала термодинамики можно записать:
| (3) |
то определение энтропии с помощью интеграла дает:
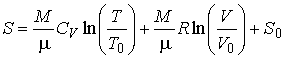 , , |
(4) |
где ![]() - произвольная постоянная интегрирования. Здесь из соображений размерности введены величины
- произвольная постоянная интегрирования. Здесь из соображений размерности введены величины ![]() и
и ![]() , которые можно считать равными единице в системе СИ:
, которые можно считать равными единице в системе СИ: ![]() К и
К и ![]() м3.
м3.
Таким образом, при ![]() энтропия, вычисленная по формуле (4), не принимает нулевого значения, а стремится к минус бесконечности. А это противоречит третьему началу термодинамики, что делает невозможным применение уравнения Клапейрона-Менделеева для описания газа при температурах, близких к абсолютному нулю. Состояние газа при
энтропия, вычисленная по формуле (4), не принимает нулевого значения, а стремится к минус бесконечности. А это противоречит третьему началу термодинамики, что делает невозможным применение уравнения Клапейрона-Менделеева для описания газа при температурах, близких к абсолютному нулю. Состояние газа при ![]() называется вырожденным состоянием и для его описания требуется применение законов, следующих из уравнений квантовой статистики.
называется вырожденным состоянием и для его описания требуется применение законов, следующих из уравнений квантовой статистики.
Список литературы
1. Савельев «Курс общей физики». Учебное пособие для ВТузов. Молекулярная физика. Первый том.
2. Кикоин «Молекулярная физика». Физмат. 1963г.
3. Т.И.Трофимова «Курс общей физики». Молекулярная физика. Лекции.
4. К.В.Глаголев «Физическая термодинамика». Том второй.
5. А.Н.Морозов. МГТУ им. Н.Б.Баумана. 2002г.