| Похожие рефераты | Скачать .docx | Скачать .pdf |
Курсовая работа: Прогрессирующие мышечные дистрофии
ГОУ ВПО Волгоградский государственный медицинский университет Росздрава
Кафедра детских болезней педиатрического факультета
Прогрессирующие мышечные дистрофии
Выполнено:
ст. 502 группы МБФ
Служенко М.О.
Проверено:
доцент Марушкин Д.В.
Волгоград, 2010
Содержание
Введение
1. Эпидемиология
2. Этиология
3. Патогенез
4. Клиническая классификация
5. Клиника
6. Диагностика
7. Лечение
8. Профилактика
Список литературы
Введение
Прогрессирующие мышечные дистрофии – это группа наследственно обусловленных нервно-мышечных заболеваний, характеризующихся прогрессирующей мышечной слабостью, атрофией мышц, двигательными нарушениями [Гусев Е.И., Никифоров А.С., 2007].
1. Эпидемиология прогрессирующих мышечных дистрофий
Первое сообщение о прогрессирующей мышечной дистрофии было опубликовано в России в 1895 г. врачом В.К. Ротом, который назвал заболевание мышечной сухоткой. Заболевание описано во всех странах мира. Частота 3,3 на 100 000 населения, 14 на 100 000 родившихся. В подавляющем большинстве случаев болеют мальчики. Случаи заболевания у девочек крайне редки, хотя и возможны при кариотипе ХО и при структурных аномалиях хромосом (Хр21.2, ген DMD дистрофина), в 35—40% случаев носит семейный характер [Гринио Л.П., Агафонов Б.В. 1997].
Частота прогрессирующей мышечной дистрофии Дюшенна варьирует от 9,7 до 32,6 на 100 000 живорожденных мальчиков. Высокая распространенность заболевания в популяции в значительной мере связана с высокой частотой новых мутаций [Bejaoui K., Hirabayashi K., Hentati F. et al., 1995].
Офтальмоплегическая мышечная дистрофия относится к числу редких заболеваний, частота встречаемости в Европе составляет 1:100 000—200 000 человек [Гусев Е.И., Коновалов А.Н., Скворцова В.И., Гехт А.Б., 2009]. Однако в некоторых этнических группах и территориальных группах с «эффектом основателя» частота ОФМД намного выше, например во франко-канадской популяции — 1:1000 человек, у евреев Бухары, — 1:600. Также описаны выборки больных с офтальмоплегической мышечной дистрофией в более чем 30 странах на всех пяти континентах [Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008].
Популяционная частота прогрессирующей мышечной дистрофии Эрба - Рота составляет 1,2-2,5 случая на 100 000 населения [Гехт Б.М. и Ильина Н.А., 1998].
Частота встречаемости плече-лопаточно-лицевой миодистрофии Ландузи — Дежерина составляет 2,9 на 100 000 населения [Яхно Н.Н., Штульмен Д.Р., Мельничук П.В., 2001].
2. Этиология
Причиной являются генетически обусловленные дефекты метаболизма или структуры мышечной ткани, приводящие к атрофии мышц, разрастанию соединительной ткани и увеличению жировой клетчатки (псевдогипертрорфии) [Гусев Е.И., Никифоров А.С., 2007].
Табл. 1 Гены, ответственные за возникновение прогрессирующих мышечных дистрофий [Nevo Y., Muntoni F., Sewery C. et al., 1998]
| Название | Английская аббревиатура, синонимы | Тип наследования | Локализация гена | Ген | Белковый продукт гена |
| ПМД Дюшенна | DMD | ХР | Xp21.2 | DMD (DYS) | Дистрофин |
| ПМД Беккера | BMD | ХР | Xp21.2 | DMD (DYS) | Дистрофин |
| ПМД Эмери-Дрейфуса со сцепленным с полом наследованием | EDMD, скапуло - перонеальная форма, плече - лопаточно - перонеальная | ХР | Xq28 | Ген Эмерина, EDMD | Эмерин |
| ПМД Эмери-Дрейфуса, аутосомно-доминантный тип | EDMD2, скапуло - перонеальная форма, плече - лопаточно - перонеальная | АД | 1q21.2 | Ген Ламина А/C (LMNA/C) | Ламин А/С |
| ПМД Эмери-Дрейфуса, аутосомно-рецессивный тип | EDMD2, скапуло - перонеальная форма, плече - лопаточно - перонеальная | АР | 1q21.2 | Ген Ламина А/C (LMNA/C) | Ламин А/С |
| ПМД Ландузи-Дежерина | FSHD1, лице - плече - лопаточная форма | АД | 4q35 | FSHMD1A | - |
| Группа конечностно-поясных ПМД | |||||
| Окулофарингеальная форма, аутосомно-доминантный тип | OPMD | АД | 14q11.2-13 | PABP2 | Полиаденилин - ассоциированный белок |
| Окулофарингеальная форма, аутосомно-рецессивный тип | OPMD | АР | 14q11.2-13 | PABP2 | Полиаденилин - ассоциированный белок |
Различные формы прогрессирующих мышечных дистрофий могут наследоваться аутосомно-доминантно, аутосомно-рецессивно, рецессивно, сцепленно с Х-хромосомой. Различные формы прогрессирующих мышечных дистрофий отличаются разным типом наследования, вариабельностью возраста начала заболевания, преимущественной локализацией поражения мышц и другими признаками [Крахмалева И.Н., Липатова Н.А., Шишкин С.С. и др., 1999].
Миодистрофии Дюшенна и Беккера являются аллельными вариантами экспрессии единого генетического дефекта в локусе Р21 Х-хромосомы. Ген является самым большим из известных на сегодняшний день и имеет очень сложную молекулярную организацию; состоит из 79 экзонов (информативно значимых участков ДНК). В 60—65 % случаев мутация представляет собой делецию гена дистрофина, а в 5—10 % — его дупликацию. Встречаются и точковые мутации гена (до 30 % случаев) [Самуэльс М., 1997]. Высокая частота спорадических случаев миодистрофий Дюшенна и Беккера обусловлена чрезвычайно высокой частотой спонтанных мутаций гена, возможно, отчасти из-за его "гигантского" размера [Свердлов Е.Д., 1997]. С локусом Р21 Х-хромосомы ассоциированы также другие, редко встречающиеся, клинические фенотипы: семейная Х-сцепленная миалгия с крампи, синдром Мак-Леода (повышение уровня КФК, акантоцитоз), квадрицепс-миопатия. Последняя является наиболее мягкой формой и характеризуется медленным профессированием слабости четырехглавых мышц бедра, гипертрофией голеней и повышением КФК. При миодистрофий Дюшенна уровень дистрофина не превышает 3 % от нормального, тогда как при болезни Беккера он колеблется от 3 до 20 % [Гехт Б.М. и Ильина Н.А., 1998].
Этиология псевдогипертрофической формы Дюшенна.
Псевдогипертрофическая прогрессирующая мышечная дистрофия (мышечная дистрофия Дюшенна, Xp21.2, ген DMD дистрофина) — возникает в результате дефектов гена, кодирующего белок дистрофин. Дистрофин локализован в плазматической мембране скелетных мышечных волокон и кардиомиоцитов. Мутации в гене дистрофина вызывают клинически явно различающиеся формы миодистрофий [Евтушенко С.К., Садеков И.А. 1994].
Некоторые авторы предполагают, что различия в клинике дистрофинопатий могут определяться характером мутаций, из которых одни приводят к сдвигу рамки считывания (в результате чего синтез соответствующего белка практически невозможен), тогда как другие повреждают ген, но не нарушают рамку считывания (результатом чего становится синтез измененного белка, частично способного к функционированию) [Евтушенко С.К., Садеков И.А. 1994]. Первый вид мутаций обычно связывают с тяжелым течением болезни, второй - с более мягким. Данное предположение, известное как гипотеза Монако, нашло подтверждение в ряде публикаций [Гринио Л.П., Агафонов Б.В., 1997].
Вместе с тем накопление сведений о мутациях в гене дистрофина и изучение клинико-генетических корреляций при дистрофинопатиях способствовали выявлению случаев, трудно объяснимых с позиций гипотезы Монако или других гипотез, постулирующих жесткую связь особенностей клиники с глубиной повреждения функции белка. Еще в 1988 г. было опубликовано сообщение о нескольких больных, у которых выявлялись делеции экзонов 3-7 гена дистрофина и соответственно сдвиг рамки считывания, но при этом заболевание протекало в мягкой форме - как миодистрофии Беккера [Шишкин С.С., 1997]. Даже если бы у пациентов только отсутствовал данный участок последовательности дистрофина (не говоря уже о нарушении рамки считывания для остальной части гена), можно было бы ожидать серьезного нарушения функции белка, так как, по имеющимся сведениям, именно эта часть молекулы дистрофина обеспечивает его связывание с актином [McKusik V., Amberger J., 2003]. Однако болезнь в ряде случаев протекала относительно доброкачественно. Более того, известен пациент с делецией экзонов 3-9 в дистрофиновом гене, который до 60-летнего возраста и не подозревал о своей болезни, а в 67 лет сохранял способность к самостоятельной ходьбе [Ahn A.H., Kunkel L.M., 1998]. Авторы, описавшие этот уникальный случай, предположили, что, следовательно, возможны делеции в функционально важной области дистрофинового гена, которые обеспечивают состояние с длительным бессимптомным течением и без существенного сокращения продолжительности жизни [Kaplan J.C., Fontaine В., 1999].
В других публикациях отмечалось, что крупные делеции, захватывающие 26% (экзоны 21-44) и даже 40% последовательности дистрофинового гена, иногда обусловливают позднее начало и очень мягкое течение болезни - такие пациенты в возрасте 55 и 60 лет сохраняли определенную двигательную активность [Шаховская Н.И., 2000].
Этиология офтальмоплегической мышечной дистрофии Кило-Невина.
В 1998 г. изолировали на хромосоме 14q11.2-13 ген поли-(А)-связывающего белка 2 (PAPB2, PABPN1), ответственный за синтез ядерного белка PABP2, служащего фактором полиаденилирования мРНК, и идентифицировали мутацию, заключающуюся в увеличении числа копий тринуклеотидных GCG-повторов в 1-м экзоне гена. В норме ген содержит шесть тандемных копий повторов GCG, а у больных их число достигает до 8—13. В некоторых популяциях экспансия числа тринуклеотидных повторов происходит за счет простого добавления GCG-повторов, в других — вместе с экспансией GCG-повторов происходит GCA-вставка.
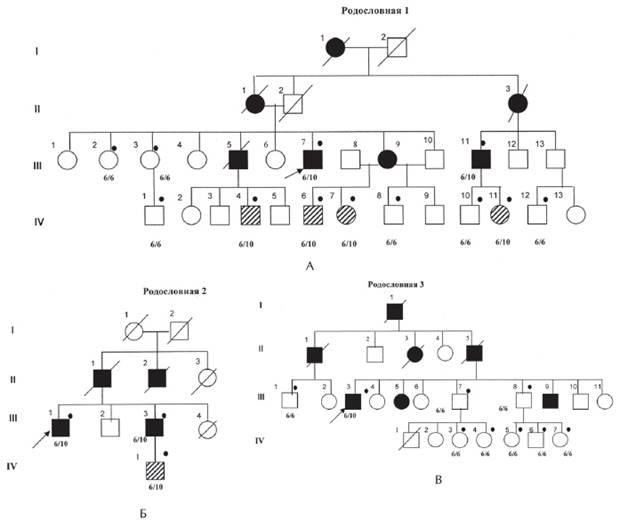
Рисунок 1. Рис. 1. А, Б, В. Родословные семей с офтальмоплегической мышечной дистрофии Кило-Невина с результатами ДНК-диагностики [Codere F., Brais B., Rouleau G., Lafontaine E., 2001].
Механизм развития экспансии тандемных повторов при офтальмоплегической мышечной дистрофии до сих пор неясен; высказываются предположения, что он может быть связан с неравным кроссинговером, разновидностью гомологичной рекомбинации, происходящей в зародышевых клетках во время мейоза или иногда митоза.
Также описаны случаи точечной мутации в гене PABPN1 22 и случаи гомозиготного носительства «промежуточного» аллеля гена, имеющего 7 GCG-повторов с аутосомно-рецессивным типом наследования с развитием более тяжелой клинической картины и более ранним дебютом симптомов заболевания [Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008].
Примечание. 6/6; 6/10 — результаты ДНК-анализа на мутацию в гене PABPN1; Закрашенный кружок —обследованные пациенты; закрашенный квадрат — больной с ОФМД, пустой квадрат — клинически здоровый; заштрихованный квадрат — клинически здоровый носитель мутации в гене PABPN1. I, II поколение — умершие родители, III поколение — больные и их сибсы, IV — дети, не достигшие возраста начала заболевания
Этиология конечно-поясной формы Эрба-Рота.
Известно не менее 9-ти локусов, ответственных за прогрессирующую мышечную дистрофию Эрба-Рота. Чаще всего вовлечен локус 15q15-q21.1 [McKusik V., Amberger J., 2003], реже вовлекается один из локусов, расположенных в коротком плече хромосомы 2 [Bashir R. et al., 1994], еще реже заболевание связывают с локусом 13q [Lim L.E., Duclos P., Broux O. et al., 1995].
В международной литературы конечностно-поясные формы обозначаются аббревиатурой LGMD (limb-girdle muscular dystrophy) c указанием типа, например LGMD 1A. Арабской цифрой 1 обозначаются типы с аутосомно-доминантным типом наследования, 2 – аутосомно-рецессивные формы [Шишкин С.С., Н.И. Шаховская, И.Н. Крахмалева, 2002]. Как видно из представленной таблицы 2, конечностно-поясные формы прогрессирующих мышечных дистрофий – это целая группа генетически гетерогенных заболеваний, объединенных общей клинической картиной: прогрессирующая проксимальная мышечная слабость и гипотрофии, симптомы «крыловидных лопаток», «утиной походки», поясничный гиперлордоз. LGMD2A соответствует ювенильной конечностно-поясной форме прогрессирующих мышечных дистрофий Эрба-Рота [Умаханова Р. С. С. Жилина Г. Р. Мутовин, 2005].
Табл.2 Гены, дефекты которых ответственны за возникновение миодистрофии Эрба-Рота (пояснично-конечностная миодистрофия или LGMD) [Умаханова Р. С. С. Жилина Г. Р. Мутовин, 2005].
| Типы | Особенности | Тип наследования | Локализация гена | Ген | Белковый продукт гена |
| 1A | АД | 5q31 | Миотилин | ||
| 1B | Аллельная форма ПМД Эмери-Дрейфуса | АД | 1q21 | Ламин А/С | |
| 1C | АД | 3p25 | CAV3 | Кавеолин-3 | |
| 1D | АД | 7q | |||
| 1E | Дилатационная кардиомиопатия | АД | 6q23 | ||
| 1F | АД | 7q32 | |||
| 1G | АД | 4p21 | |||
| 2A (Эрба) | Начало 2-45 лет, в среднем 14-20 лет. | АР | 15q15.1-q15.3 | CAPN3 | Кальпаин-3 |
| 2B | Аллельная форма – дистальная дистрофия Миоши | АР | 2p13.1 | Дисферлин | |
| 2C | АР | 13q12 | SGCG | ||
| 2D | АР | 17q21 | SGCA | ||
| 2E | АР | 4q12 | SGCB | ||
| 2F | АР | 5q33 | SGCD | ||
| 2G | АР | 17q11-12 | Телетонин | ||
| 2H | АР | 9q31-33 | TRIM32 | ||
| 2I | Аллельная форма мерозиновой миопатии (ламинин-2) и конгенитальной мышечной дистрофией с мышечными гипертрофиями и нормальной ЦНС | АР | 19q13.3 | FKRP | Фукутин-связанный белок |
| 2J | Аллельная форма дилатационной кардиомиопатии 1G и Finnish дистальной миопатии | АР | 2q31 | Титин | |
| 2K | С умственной отсталостью | АР | 9q34 | POMT1 |
Экспрессивность генов конечно-поясной формы Эрба-Рота значительно варьирует не только в популяции, но даже в пределах одной пораженной семьи, что, по-видимому, и определяет различную тяжесть и прогрессирование миодистрофического процесса у больных, а также существование относительно доброкачественных или злокачественных форм патологии [Бадалян Л.О., 2008; Вельтищев Ю.Е. и соавт., 1998].
Этиология миодистрофии Дрейфуса-Хогана
В 1990-х годах последовательно были идентифицированы ген эмерина в локусе Хq28, ответственный за Х-сцепленную миодистрофию Дрейфуса-Хогана, и ген ламинов LMNA в локусе 1q21.2, вызывающий аутосомно-доминантную форму, вслед за чем появились многочисленные верифицированные наблюдения обоих генетических вариантов [Бадалян Л. О., Темин П.А., Калинин В.А. и др., 1990; Руденская Г.Е., Тверская С.М., Чухрова А.Л. и др., 2004].
Клиническая общность Х-сцепленной и аутосомно-доминантной форм неслучайна: она обусловлена тесным функциональным взаимодействием ламинов А/С и эмерина - мембранных белков, участвующих в образовании каркаса ядерной оболочки. При общем основном симптомокомплексе клиническая картина обеих генетических форм миодистрофии Дрейфуса-Хогана, особенно аутосомно-доминантной, демонстрирует разнообразие по возрасту начала, темпам прогрессирования, выраженности отдельных симптомов и тяжести болезни в целом, активности КФК, наличию атипичных признаков. Меж- и внутрисемейное клиническое разнообразие отмечалось уже в первых описаниях Х-сцепленной и аутосомно-доминантной миодистрофии Дрейфуса-Хогана вариантов [Бадалян Л. О., Темин П.А., Калинин В.А. и др., 1990].
В международных базах данных зарегистрировано более 100 мутаций гена эмерина и около 200 мутаций LMNA (большинство - при миодистрофии Дрейфуса-Хогана), действительное число мутаций несомненно больше, поскольку не все исследователи регистрируют свои находки в базах данных. Преобладают миссенс-мутации. Преобладающих по частоте («мажорных») мутаций нет, большинство мутаций обоих генов встречаются в единичных семьях, но некоторые описаны неоднократно. К ним относится мутация Arg249Gln в экзоне 4 гена LMNA, выявленная у больных с разными фенотипами. Мутация Arg249Gln возникает de novo, что позволяет предполагать наличие мутационной «горячей точки» в гене LMNA [Руденская Г.Е., Тверская С.М., Чухрова А.Л. и др., 2004]. Так же имеет место мутация Arg377His в экзоне 6 [Мальмберг С.А., Петрухин А. С., Широкова В.И., 2000].
Нормальный биохимический продукт гена эмерин – представляет собой обогащенный аминокислотой (серином) белок, состоящий из 254 аминокислот. Эмерин экспрессируется преимущественно в скелетных, гладких мышцах и кардиомиоцитах; ему принадлежит значительная роль в организации клеточного цитоскелета и везикулярного транспорта. В сердечной мышце эмерин обеспечивает межклеточную адгезию и осуществление контактов между кардиомиоцитами. Типичная мутация представлена делецией гена и приводит к прекращению синтеза эмерина [Крахмалева И.Н., Липатова Н.А., Шишкин С.С. и др., 1999].
Этиология миодистрофии Бетлема
Редкая доброкачественная миодистрофия, наследуемая по аутосомно-доминантному типу. Установлена генетическая гетерогенность болезни: один из генов картирован в локусе 21q22, другой — 2q37. В результате мутаций нарушается синтез субъединиц коллагена VI типа, который обеспечивает связь базальной мембраны с гликопротеинами внеклеточного матрикса [Вельтищев Ю.Е., Темин П.А., 1998].
3. Патогенез
Существует несколько гипотез патогенеза прогрессирующих мышечных дистрофий. К настоящему времени точно установлено, что важным патогенетическим звеном является повышенная проницаемость мембран мышечных клеток [Евтушенко С.К., Садеков И.А. 1994]. Имеются также данные, прямо или косвенно указывающие на существование мембранного дефекта при других прогрессирующих мышечных дистрофиях [Иллариошкин С.Н., Иванова-Смоленская И.А.. 1998]. В частности, к свидетельствам повреждения мембран относят многократное повышение содержания в крови пациентов ряда мышечных ферментов и других мышечных белков (креатинфосфокиназы, трансаминаз). Наряду с этим отмечается, что существенную роль в развитии дистрофического процесса при ПМ могут играть нарушения обмена Са2+, приводящие к повышению его концентрации в цитоплазме клеток и активации Са2+-зависимых нейтральных протеиназ, которые в свою очередь запускают процессы разрушения мышечных белков [Горбунова В.Н., Савельева Е.А., Красильников В.В., 2000]. Обсуждается также гипотеза об участии активных форм кислорода и свободных радикалов в запуске механизмов клеточной гибели при прогрессирующих мышечных дистрофиях. Однако принципиально важным представляется то, что мембраны мышечных клеток при прогрессирующих мышечных дистрофиях становятся проницаемыми для многих внутриклеточных белков и эти белки из клеток попадают в кровь [Баранов В.С., 1999].
По всей видимости, мышечные белки в крови могут восприниматься иммунной системой организма как чужеродный материал, и тогда на него будет возникать иммунный ответ, усиливающийся с возрастом пациента. В принципе такой иммунный ответ должен вести ко вторичному повреждению мембран мышечных клеток и еще больше усиливать выход в кровоток мышечных белков. Тем самым, возможно, определяется прогрессирование болезни. По крайней мере роль подобных аутоиммунных механизмов отмечена в патогенезе уже целого ряда заболеваний [Горбунова В.Н., Савельева Е.А., Красильников В.В., 2000].
Более того, обычно в мышцах пациента с миодистрофией Дюшенна наблюдаются разрастание соединительной ткани, инфильтрация лимфоцитами и жировое перерождение мышечных волокон. Все это в какой-то степени сходно с морфологической картиной других аутоиммунных заболеваний [Иллариошкин С.Н., Иванова-Смоленская И.А.. 1998].
Определенным подтверждением данного механизма патогенеза прогрессирующих мышечных дистрофий является успешное использование иммунодепрессантов с целью ослабления иммунного ответа организма на белки, поступающие в кровоток пациента из мышц, охваченных дистрофическим процессом [Шишкин С.С., Калинин В.Н., 1999].
Показано, что имеют место нарушения многих биохимических констант, различные электрофизиологические и ультраструктурные изменения. Определенную роль в развитии патологического процесса при прогрессирующих мышечных дистрофиях играет синтез неполноценных мышечных белков актина и миозина, сопровождающийся их ускоренным распадом. Обнаружены нарушения активности ряда неспецифических ферментов (креатинфосфокиназы, альдолазы и др.). Выявлены нарушения энергетического обмена, выражающиеся в быстром распаде соединений, используемых в качестве энергетических ресурсов при сокращении мышц. Определенную роль в развитии патологического процесса играет нарушение строения клеточных мембран, приводящее к изменению их проницаемости для ионов калия, натрия. Эти ионы имеют значение для сокращения мышц. В развитии дистрофии мышц определенная роль принадлежит патологии капилляров и нарушениям строения соединительной ткани [Liu J., Aoki M., Illa I. et al., 1998].
При прогрессирующих мышечных дистрофиях основной патологический процесс развивается в мышечной ткани; при другой группе болезней изменения в мышцах возникают вторично, первично нарушается структура нервной клетки и волокна. Эти заболевания носят название неврогенных мышечных атрофии. К ним относят спинальные (протекающие с преимущественным поражением двигательных клеток спинного мозга) и невральные (с поражением периферических нервов) амиотрофии [McNally E., Passos-Bueno R., Bonnemann C.G. et al., 1996].
В группу прогрессирующих мышечных дистрофий относят заболевания, различающиеся по времени появления клинических симптомов, преимущественной локализации мышечных атрофии, характеру их распространения, темпу нарастания патологических изменений и типу наследования [Шишкин С.С., 1998].
Основные патоморфологические изменения при прогрессирующих мышечных дистрофиях находят в мышцах. Они выражаются в атрофии отдельных мышечных волокон. Миофибриллы утрачивают поперечную исчерчешюсть, а иногда и полностью разрушаются. В ядрах мышечных клеток также обнаруживают изменения. Они становятся крупнее обычных, содержат различные включения, иногда сморщиваются. На месте атрофированных волокон интенсивно разрастается жировая и соединительная ткань. Нервные волокна и нервные клетки остаются относительно сохранными. Выраженные изменения находят в сосудах мышц, в которых имеется тенденция к сужению и образованию тромбов [Иллариошкин С.Н., Иванова-Смоленская И.А.. 1998].
4. Клиническая классификация [Казаков В.М., 2001]
| Тип | Генетический механизм | Клинические признаки | Вовлечение других систем органов | ||
| Дюшенна | Х-хромосомная рецессивная мутация дистрофин-гена | Начало в возрасте до 5 лет; прогрессирующая слабость мышц тазового и плечевого пояса; неспособность ходить после 12 лет; кифосколиоз; дыхательная недостаточность в возрасте 20-30 лет | Кардиомиопатия; снижение интеллекта | ||
| Беккера | Х-хромосомная рецессивная мутация дистрофин-гена | Начало в раннем или позднем возрасте; медленно прогрессирующая слабость мышц тазового и плечевого пояса; сохранение способности ходить после 15 лет; дыхательная недостаточность после 40 лет | Кардиомиопатия | ||
| Миотоническая | Аутосомно- доминантный; расширение нестабильного участка ДНК хромосомы 19ql3,3 | Начало в любом возрасте; медленно прогрессирующая слабость мышц век, лица, шеи, дистальных мышц конечностей; миотония | Нарушение сердечной проводимости; психические нарушения; катаракты, лобная алопеция; атрофия гонад | ||
| Плече-лопаточно-лицевая | Аутосомно-доминантный; часто мутации хромосомы 4q35 | Начало в возрасте до 20 лет; медленно прогрессирующая мышечная слабость лицевой области, плечевого пояса, тыльного сгибания стопы | Гипертензия; глухота | ||
| Плечевого и тазового пояса (возможны несколько заболеваний) | Аутосомно-рецессивный или доминантный | Начало с раннего детства до среднего возраста; медленно прогрессирующая слабость мышц плечевого и тазового пояса | Кардиомиопатия | ||
| Глазо-глоточная | Аутосомно-доминантный (Французская Канада или Испания) | Начало в 50-60 лет; медленно прогрессирующая слабость мышц: наружных глазных, век, лица и глотки; крикофарингеальная ахалазия. | Церебральные, глазные | ||
| Врожденная (включает несколько заболеваний, в том числе типы Фукуяма ицеребро-окулярная дисплазия) | Аутосомно-рецессивный | Начало при рождении; гипотония, контрактуры, задержка развития; в одних случаях — ранняя дыхательная недостаточность, в других — более благоприятное течение болезни | |||
| Основные выявленные клинические варианты миодистрофии Дюшена (МДД) | |||||
| Вариант МДД | Возраст начала болезни, годы | Способность к ходьбе и состояние опорно-двигательной системы | Масса тела, интеллект, осложнения | Число случаев и процент от общего числа пациентов | |
| I (классическое течение) | 2-5 | Теряет способность ходить в 10-12 лет. Генерализованная мышечная слабость, затем сколиоз, контрактуры голеностопных, коленных и других суставов | Масса тела снижена. Психическое развитие в норме; кардиомиопатия обнаруживается после 8-10 лет | 62(30,2%) | |
| II (с кушингоидным синдромом) | 2-5 | Теряет способность ходить в 10 лет или ранее. Генерализованная мышечная слабость, затем сколиоз, контрактуры голеностопных и других суставов | Ожирение (лунообразное лицо, отложение жира по женскому типу). Kардиомиопатия обнаруживается после 10 лет | 44(21,5%) | |
| III (врожденная форма) | 1-2-ой год жизни | Теряет способность ходить до 10 лет, иногда в 6,5-7 лет. Ранние множественные контрактуры. Быстрое прогрессирование | Масса тела снижена или в норме. Задержка психического развития. Kардиомиопатия обнаруживается в 7-10 лет | 28(13,7%) | |
| IV (кардиомиопатический) | 2-6 | В 6,5-7 лет обнаруживается кардиомиопатия при небольших проявлениях мышечной слабости (затруднения при подъеме по лестнице). Относительно медленное прогрессирование | Масса тела снижена или в норме. Психическое развитие в норме | 15(7,3%) | |
| V (смешанный) | 1-6 | Теряет способность ходить в 10-12 лет или ранее. Генерализованная мышечная слабость | Различные сочетания | 56(27,3%) | |
5. Клиника
Первые признаки болезни проявляются нарастающей слабостью тех или иных групп мышц, утомляемостью при легких физических нагрузках, симметричными атрофиями мышц [Евтушенко С.К., Садеков И.А. 1994].
Характерными симптомами прогрессирующих мышечных дистрофий являются мышечная слабость и атрофия мышц, которые могут проявляться в различные возрастные периоды, но чаще развиваются в детском и юношеском возрасте [Гаусманова - Петрусевич И., 2001]. Дети поздно начинают ходить, быстро утомляются, неуклюжи в ходьбе, спотыкаются при беге, часто падают, с трудом поднимаются по лестнице. Двигательные нарушения постепенно прогрессируют. Возникает миопатическая утиная походка. В случае поражения мышц тазового пояса и конечностей затруднен переход из горизонтального положения в вертикальное; при поражении дистальных групп мышц ног появляется петушиная походка. Стойкость и нарастание двигательных нарушений позволяют диагностировать миодистрофию уже на ранних стадиях заболеваниях. При обследовании больного обнаруживают генерализованную или локальную атрофию мышц. Локальная атрофия мышц выявляется лишь на ранних стадиях заболевания, по мере прогрессирования патологического процесса атрофия мышц приобретает генерализованный характер вплоть до мышечной кахексии. Атрофированные мышцы истончены, дряблые при пальпации, однако следует отметить, что наряду с атрофией мышц выявляется псевдогипертрофия (замещение атрофированных мышц жировой клетчаткой, соединительной тканью). Миодистрофический процесс сопровождается поражением соединительной ткани, миосклерозом, развитием сухожильно-связочных ретракций, ограничением объема движений в суставах, укорочением пяточного (ахиллова) сухожилия, контрактурами. Одновременно с развитием мышечных атрофий снижаются сухожильные рефлексы, в первую очередь коленные [Вельтищев Ю.Е., Темин П.А., 1998].
Поражение мышц плечевого пояса приводит к ограничению движений в плечевых суставах. Больные не могут поднять руки выше горизонтального уровня, в то время как объем движений в локтевых и лучезапястных суставах и сила мышц длительное время остаются сохранными. При попытке поднять больного подмышки его голова как бы проваливается в плечи — симптом «свободных надплечий». Лопатки отстают от туловища — симптом «крыловидных лопаток». При поражении мышц тазового пояса возникают затруднения при подъеме на лестницу, вставании из положения сидя. При этом больной оказывает себе помощь, опираясь на посторонние предметы, встает в несколько этапов («лесенкой»). Изменяется походка: она становится переваливающейся, раскачивающейся - «утиная» походка. Атрофия косых мышц живота приводит к развитию «осиной» талии. Слабость длинных мышц спины нарушает осанку, приводит к искривлению позвоночника и выпячиванию живота [Гаусманова - Петрусевич И., 2001].
Поражение мышц костей и стоп сопровождается их слабостью. Походка больных становится своеобразной. Для того чтобы не зацепиться носком отвисающей стопы за пол, больные вынуждены высоко поднимать голень— «петушиная» походка [Евтушенко С.К., Садеков И.А., 1994].
При слабости и атрофии мышц лица отмечается отсутствие морщин на лбу (симптом «полированного лба»). Наблюдается гипомимия: больные не могут плотно зажмурить глаза, надуть щеки, вытянуть губы в трубочку и т. д. В некоторых случаях вследствие замещения губных мышц соединительной и жировой тканью губы утолщаются (напоминают губы тапира) [Hoffmann E.P., Kunkel L.M., Angelini C. et al., 2009].
При поражении наружных глазных мышц отмечается ограничение объема движения глазных яблок; иногда они становятся полностью неподвижными [McNally E., Passos-Bueno R., Bonnemann C.G. et al., 1996].
Если в патологический процесс вовлекаются мышцы глотки и гортани, возникает осиплость голоса и нарушается акт глотания. Поражение межреберных мышц ведет к дыхательной недостаточности и заболеваниям легких и сердца [Minetti C., Sotgia F., Bruno C. et al., 1998].
При неврологическом обследовании больных с прогрессирующими мышечными дистрофиями наряду с ограничением объема движений, снижением силы мышц и их атрофией выявляются мышечная гипотония, снижение или полное отсутствие сухожильных рефлексов [Moreira E., Vainzof M., Marie S. et al., 1997].
Темп прогрессирования патологического процесса зависит от формы заболевания и индивидуальных особенностей организма. В стадии выраженных нарушений вследствие атрофии мышц и отсутствия движений могут формироваться контрактуры (тугоподвижность или невозможность движения в суставах) [Minetti C., Sotgia F., Bruno C. et al., 1998].
Большинство форм прогрессирующих мышечных дистрофий не сопровождается снижением интеллекта. Больные критически относятся к своему дефекту. Иногда наблюдаются выраженные эмоциональные нарушения в виде повышенной раздражительности, подавленности настроения, замкнутости. Большинство больных успешно обучается по программе массовой школы [Muntoni F., Mateddu A., Marchei F. et al., 1993].
Исключение составляют больные псевдогипертрофической формой. При этой форме наблюдается выраженное снижение интеллекта. Данный вариант прогрессирующей мышечной дистрофии наследуется рецессивно, сцеплен-но с У-хромосомой. Основную массу больных составляют мальчики. Наряду с прогрессирующими атрофиями и слабостью мышц плечевого и тазового пояса у больных наблюдаются псевдогипертрофии (разрастания соединительной ткани, особенно в области икроножных мышц) и эндокринные нарушения (чаще ожирение). Некоторая задержка развития психических функций отмечается уже в первые годы жизни. Дети малоэмоциональны. Речь развивается с запозданием и носит примитивный характер. Отсутствует абстрактное мышление. Навыки опрятности и самообслуживания формируются с трудом. Интеллект обычно классифицируется как тяжелая дебильность или имбецильность; реже наблюдается идиотия [Гаусманова - Петрусевич И., 2001].
Псевдогипертрофическая злокачественная миодистрофия Дюшенна

Первое наблюдение псевдогипертрофической миопатии принадлежит E.Meryon (1852), опубликовавшему в статье "К вопросу о жировой и гранулярной дегенерации мышц" семейный случай болезни у 4 братьев с аномальным увеличением икроножных мышц и контрактурами конечностей. G.Duchenne в 1861 г. описал больного с "псевдогипертрофическим мышечным параличом", обратив внимание на необычное сочетание увеличения икроножных мышц с прогрессирующей мышечной слабостью. В 1868 г. G.Duchenne опубликовал серию статей в журнале "Архив общей медицины", где представил систематизированный анализ болезни [Moreira E., Vainzof M., Marie S. et al., 1997].
Проявляется в возрасте 2—5 лет. Течение быстро прогрессирующее, злокачественное. Обездвиженность больных, как правило, наступает в возрасте 14—15 лет, смерть наступает в возрасте 15—18 лет, больные редко живут более 25 лет. К 8-10 годам большинство детей нуждается в ортопедических аппаратах; к 12 годам большинство детей не могут ходить. Первые признаки заболевания проявляются в 1-3 года жизни слабостью мышц тазового пояса. Уже на 1-м году обращает на себя внимание отставание детей в моторном развитии. Они, как правило, с задержкой начинают садиться, вставать, ходить. Движения неловкие, при ходьбе дети неустойчивы, часто спотыкаются, падают. В 2-3 года появляются мышечная слабость, патологическая мышечная утомляемость, проявляющаяся при физической нагрузке - длительной ходьбе, подъеме на лестницу, изменение походки по типу «утиной». В этот период обращает на себя внимание своеобразная «стереотипная» динамика движений детей во время вставания из горизонтального положения, с положения на корточках или со стула. Вставание происходит поэтапно с активным использованием рук - «взбирание лесенкой» или «взбирание по самому себе». Типичные жалобы родителей — это ходьба детей на пальцах и частые падения. Задержка темпов двигательного развития часто обнаруживается ретроспективно при анализе анамнестических сведений. Ранние симптомы подкрадываются незаметно. Недостаточную, по сравнению со сверстниками, подвижность ребенка, его двигательную пассивность часто относят к особенностям темперамента и характера [Гринио Л.П,. 1998].
Псевдогипертрофия икроножных мышц создает обманчивое впечатление о сохранности мышечной силы и даже радует родителей. Псевдогипертрофии мышц могут развиваться также в ягодичных, дельтовидных мышцах, мышцах живота и языка. Дети могут не привлекать внимания специалиста до тех пор, пока проксимальная мышечная слабость не станет настолько выраженной, что затруднит вставание ребенка с пола и определит утиный тип ходьбы и использование миопатических приемов "взбирания по себе" (симптом Говерса). Ретракция пяточных (ахилловых) сухожилий не позволяет больному полноценно опираться на пятки, что определяет ходьбу на пальцах. На протяжении детства двигательная сила постепенно снижается. Двигательные функции выглядят относительно стабильными между 3 и 6 годами жизни. В большинстве случаев возможность ходьбы и подъема по лестнице сохраняется до 8-летнего возраста. Между 3 и 8 годами происходит нарастающее укорочение пяточных сухожилий и формируются сгибательные контрактуры в голеностопных суставах, развиваются поясничный гиперлордоз, кифосколиоз грудного отдела позвоночника. Нарастают атрофии мышц бедра, тазового пояса, а затем плечевого пояса, спины и проксимальных отделов рук. Атрофии мышц всегда симметричны. Нередко атрофии мышц маскируются хорошо развитой подкожной жировой клетчаткой. Изменения костной системы не ограничиваются лишь сколиозом: часто развиваются деформации грудной клетки и стоп, диффузный остеопороз. Ухудшение походки ведет к тому, что дети все чаще падают. Вначале атрофии локализуются в проксимальных группах мышц нижних конечностей - мышцах тазового пояса, бедер, а через 1-3 года быстро распространяются в восходящем направлении на проксимальные группы мышц верхних конечностей - плечевой пояс, мышцы спины. Вследствие атрофии появляются лордоз, «крыловидные» лопатки, «осиная» талия. Типичным, «классическим» симптомом заболевания является псевдогипертрофия икроножных мышц. При пальпации мышцы плотные, безболезненны. У многих больных в результате селективного и неравномерного поражения различных групп мышц рано возникают мышечные контрактуры и сухожильные ретракции. Мышечный тонус снижен преимущественно в проксимальных группах мышц. Глубокие рефлексы изменяются с различной последовательностью. В ранних стадиях болезни исчезают коленные рефлексы, позже - рефлексы с двуглавой и трехглавой мышц. Ахилловы рефлексы длительное время остаются сохранными. Характерны симметричная и неуклонно прогрессирующая слабость в мышцах бедер и плечевого пояса, затрудняющая движения при подъеме, беге, прыжках, поясно-конечностная атрофия мышц, преимущественно мышц тазового пояса и бедер, истинная гипертрофия или псевдогипертрофия икроножных мышц, ранние сухожильно-связочные ретракции (укорочение сухожилий и связок), контрактуры крупных суставов. Коленные рефлексы рано исчезают, ахилловы рефлексы сохраняются [Muntoni F., Mateddu A., Marchei F. et al., 1993].
Одной из отличительных особенностей миодистрофии Дюшенна является сочетание данной формы с патологией костно-суставной системы и внутренних органов (сердечно-сосудистой и нейроэндокринной систем). Костно-суставные нарушения характеризуются деформациями позвоночника, стоп, грудины. На рентгенограммах обнаруживают сужение костномозгового канала, истончение коркового слоя длинных диафизов трубчатых костей [Novakovic I., Todorovic S., Apostolski S. et al., 1998].
Сердечно-сосудистые расстройства клинически проявляются лабильностью пульса, артериального давления, иногда глухостью тонов и расширением границ сердца. На ЭКГ регистрируются изменения миокарда (блокада ножек пучка Гиса и др.). Нейроэндокринные нарушения встречаются почти у половины пациентов. Чаще других даются синдром Иценко-Кушинга, адипозогенитальная дистрофия Бабинского-Фрелиха [Страхова О.С., Белозерова Ю.М., Темин П.А., 1999].
Установлено, что при мышечной дистрофии Дюшенна сердечно-сосудистая система вовлекается в патологический процесс достаточно часто и рано. Около 73% больных с данной нозологией имеют различные проявления кардиальной патологии. Причиной сердечно– сосудистой патологии является генетически детерминированный недостаток дистрофина в кардиомиоцитах [Страхова О.С., Белозерова Ю.М., Темин П.А., 1999].
Отсутствие четкой корреляции между тяжестью поражения скелетных мышц и наличием выраженной кардиомиопатии у пациентов с прогрессирующей мышечной дистрофии Дюшенна предопределило необходимость обратить особое внимание на исследование маркеров вовлечения сердечной мышцы в патологический процесс. Оказалось, что делеции гена дистрофина являются не единственной причиной поражения мышечной ткани у пациентов с прогрессирующей мышечной дистрофии Дюшенна. В настоящее время ученые выделяют три основных причины: дефицит дистрофина, обусловленный генетическим дефектом; дефицит дистрофин- ассоциированного гликопротеина (молекулярная масса 50 кДа) или других дистрофинассоциированных белков, наличие особого генетического варианта строения ангиотензин- конвертирующего фермента. Сердечная мышца может поражаться как вследствие всех трех причин, так и их комбинаций. Например, дефицит дистрофинассоциированного гликопротеина у пациентов с прогрессирующей мышечной дистрофией Дюшенна может наблюдаться исключительно в кардиомиоцитах, в то время как в склетной мышечной ткани его содержание будет нормальным . Обнаружение дефицита дистрофинассоциированных белков при исследовании биоптата сердечной мышцы является предиктором развития тяжелой кардиомиопатии. Особое внимание последние годы уделяется строению ангиотензин-конвертирующего фермента. По мнению Kasper EK с соавт. тяжесть кардиомиопатии при прогрессирующей мышечной дистрофии Дюшенна взаимосвязана особенностями строения ангиотензин- конвертирующего фермента у больного. Выявление маркеров вовлечения сердечной мышцы в патологический процесс позволяет ответить на исключительно важный практический вопрос – почему кардиомиопатия может наблюдаться у пациентов с легкими вариантами поражения скелетных мышц, а также возможность дебюта заболевания с кардиомиопатии [Nigro V., de Sa Moreira E., Piluso G. et al., 1996].
По данным А.Oldfors, начальные проявления кардиальной патологии у больных возникают уже в раннем возрасте и прогрессируют с годами. В отдельных случаях, у детей 3-5 лет, в клинической картине заболевания могут преобладать кардиальные симптомы, а симптомы мышечной дистрофии могут быть маскированными [Novakovic I., Todorovic S., Apostolski S. et al., 1998].
Низкая физическая активность пациентов с прогрессирующей мышечной дистрофии Дюшенна, относительно быстрая утрата способности к самостоятельной ходьбе, снижающая нагрузку на миокард, а также недостаточная нацеленность родителей на выявление кардиальных жалоб (основное внимание обращается прежде всего на двигательные нарушения), приводят к тому, что менее 15% детей до 14 лет, имеющих поражение мышцы сердца, активно обращаются к кардиологу. В то время как по данным целевых исследований у детей, не предъявляющих кардиальных жалоб, поражение мышцы сердца выявляется у 25% в возрасте до 6 лет и у 59% в возрасте от 6 до 10 лет. В дальнейшем этот процент снижается, поскольку поражение сердца прогрессирует и дети начинают предъявлять кардиальные жалобы [Nigro V., de Sa Moreira E., Piluso G. et al., 1996].
Патогенез поражения мышцы сердца при прогрессирующей мышечной дистрофии Дюшенна в настоящее время представляется следующим образом [Ishikawa Y, Bach JR, Sarma RJ et al., 1995]: прогрессирующая атрофия кардиомиоцитов и замещение их фиброзной тканью приводят к истончению миокарда (особенно левого желудочка, на который приходится основная гемодинамическая нагрузка), а также к снижению его способности к систолическому сокращению и диастолическому расслаблению. Выраженный фиброз в области задних папиллярных мышц ведет к пролабированию створок митрального клапана в полость левого предсердия (пролапс митрального клапана) с или без наличия митральной регургитации. Частота выявления пролапса митрального клапана у пациентов с прогрессирующей мышечной дистрофии Дюшенна составляет от 25 до 55% [Ishikawa Y, Bach JR, Sarma RJ et al., 1995]. Увеличение размеров левого предсердия как правило вторично, вследствие митральной регургитации или снижения сократительной способности левого желудочка. Нарушения ритма сердца и проводимости возникают вследствие прогрессирующего фиброза проводящей системы сердца [Novakovic I., Todorovic S., Apostolski S. et al., 1998].
Обычно поражение сердечной мышцы впервые диагностируется между 6 и 7 годами. С возрастом частота выявления кардиальных симптомов возрастает, и к 20 годам патология сердечно- сосудистой системы встречается у 95% больных. Наиболее частыми нарушениями, наблюдавшимися у 54% пациентов, были: тахикардия, аритмии и сердечная недостаточность. Особенно выражены данные симптомы в конечных стадиях заболевания [Adzija D et al.,1994].
Учитывая особенности двигательной активности пациентов с прогрессирующей мышечной дистрофией Дюшенна (а также с миодистрофией Бекера), отсутствие кардиальных жалоб, весьма малую физическую активность (как правило, больные к 10-11 годам теряют способность к самостоятельной ходьбе), часто обездвиженность на поздних этапах заболевания, G. Nigro с соавторами в 1993 году был предложен и введен в медицинскую практику новый диагностический термин – латентная сердечная недостаточность [Nigro V., de Sa Moreira E., Piluso G. et al., 1996].
Так же особенностями этой формы прогрессирующей мышечной дистрофии являются сопутствующая поражению мышц умственная отсталость, снижение интеллекта, остеопороз и истончение кортикального вещества костей, кардиомиопатия, легочно-сердечная недостаточность. У части больных обнаруживаются различные признаки эндокрпинопатии: адипозогенитальный синдром, низкорослость. В связи с дефицитом церебральных изоформ дистрофина — аподистрофинов, у 30 % больных с миодистрофией Дюшенна имеет место умственная отсталость различной степени: от пограничной интеллектуальной недостаточности до выраженной олигофрении. Тяжесть олигофрении и нарушений высших когнитивных функций не коррелирует с выраженностью мышечного дефекта и стадией миодистрофического процесса. К экзогенным факторам, усугубляющим проявления умственной отсталости, относят развивающуюся социальную дезадаптацию вследствие невозможности из-за двигательного дефекта полноценного участия детей в детских коллективах (сад, школа), влияние неблагоприятных перинатальных причин и, возможно, дисгенезий головного мозга (при КТ и МРТ изредка обнаруживают признаки церебральной атрофии) [Гринио Л.П., 1998].
Сопутствующие нарушения. Сухожильные и мышечные контрактуры (в том числе ахилловых сухожилий), прогрессирующий кифосколиоз, нарушение функции легких, кардиомиопатия, интеллект снижен. Мышечная слабость сочетается с пальпаторно определяемым увеличением и плотностью некоторых мышц (например, икроножных), что вначале является результатом гипертрофии, а затем замещения мышц жировой и соединительной тканью [Гринио Л.П., 1998].
Слабость дыхательной мускулатуры и диафрагмы обусловливает уменьшение ЖЕЛ до 20 % от нормы, что приводит к эпизодам ночной гиповентиляции. Дети часто встают со страхом, связанным с ощущением удушья, и боятся спать. Существенный вклад в летальность вносит дыхательная недостаточность, которая провоцируется интеркуррентными инфекциями или аспирацией [Тетенев Ф.Ф., Бодрова Т.Н., Емельянова Н.В., 2000].
Псевдогипертрофическая доброкачественная миодистрофия Беккера — Кинера.
Второй по частоте Х-сцепленной формой является так называемая доброкачественная форма псевдогипертрофической миодистрофии Беккера — Кинера. Впервые доброкачественная форма была описана в 1955 г. P . Becker и F . Kiener . В последующем V. McKu — sick (1964), R . Shaw , F . Dreifuss (1969) сообщили о подобном заболевании как самостоятельной мутации [Евтушенко С.К., Садеков И.А. 1994].
Менее тяжелая и реже встречающаяся, чем дистрофия Дюшенна, с более медленным течением и более поздним началом, но со сходными клиническими и лабораторными признаками. Это заболевание также является результатом дефекта в дистрофин-гене. Начинается в возрасте 10—15 лет. Поражаются мышцы проксимальных отделов конечностей, тазового и плечевого пояса [Bushby K.M.D. et al, 1993].

Течение сравнительно мягкое, длительное время сохраняется возможность самообслуживания и даже трудоспособность. В отличие от формы Дюшенна не наблюдается нарушений интеллекта. По топографии мышечного поражения миодистрофия Беккера—Кинера полностью повторяет миодистрофию Дюшенна. Слабость и гипотрофия вначале проявляются в мышцах тазового пояса и бедер, затем процесс распространяется на мышцы плечевого пояса. Мышцы лица обычно интактны. Псевдогипертрофии развиваются в икроножных мышцах почти во всех случаях, могут быть очень значительными, иногда они отмечаются и в других мышечных группах. Наблюдается прогрессирующий поясничный лордоз, появляется утиная походка, затруднение при подъеме с пола ("приемы миопата"), беге, а в поздних стадиях и при ходьбе. Развиваются сухожильные ретракции, в первую очередь в ахилловых сухожилиях. Кардиомиопатня при форме Беккера—Кинера или совсем не встречается или выражена очень слабо. При миодистрофий Беккера, как и при форме Дюшенна, повышен уровень креатинфосфокиназной активности, однако незначительно [Le-Thiet-Thanh; Nguyen-Thi-Man; Hori-S; Sewry-CA; Dubowitz-V, 1995].
До настоящего времени окончательно не решен вопрос о том, являются ли обе формы миодистрофий, сцепленные с Х-хромосомой, самостоятельными нозологическими формами или это разновидности течения одной болезни. Следует учесть, что при миодистрофий Беккера имеется сцепление с цветовой слепотой (иногда лишь частичной), что не встречается при форме Дюшенна. Не наблюдается благоприятного течения при экспериментальной миодистрофий у животных [Khurana T.S., Prendergast R.A., Alameddine H. et al., 1995]. С практической точки зрения, учитывая различный прогноз, следует обязательно различать эти две формы [Maeda M; Nakao S; Miyazato H; et al., 1996].
Электромиографические, биохимические и патоморфологические изменения умеренно выражены, отмечается изменение цветового зрения. Длительное время у больных сохраняются трудоспособность, возможность самостоятельного передвижения, интеллект; кардиомиопатия выражена умеренно [Maeda M; Nakao S; Miyazato H; et al., 1996].
Миодистрофия Дрейфуса — Хогана.
Мышечная дистрофия Дрейфуса— Хогана известна с конца 1960-х годов, когда была описана первая семья с Х-сцепленной формой болезни. Позднее наряду с другими описаниями этой формы появились единичные сообщения о клинически неотличимой миодистрофии с аутосомно-доминантным типом наследования. На протяжении нескольких десятилетий оба варианта, особенно аутосомно-доминантный, считались очень редкими. С развитием ДНК-диагностики эти представления изменились. Оказалось, что мышечная дистрофия Дрейфуса-Хогана вносит значимый вклад в структуру мышечных дистрофий. Относится к редким Х-хромосомным формам прогрессирующих мышечных дистрофий. Мышечная дистрофия Дрейфуса- Хогана является медленно прогрессирующей формой миодистрофии с Х-сцепленным рецессивным типом наследования [Белозеров Ю.М., Никанорова М.Ю., Перминов В.С., Страхова О.С., 2001].
Заболевание дебютирует между 5 и 15 годами жизни. Самыми ранними и типичными признаками обычно являются развивающиеся сгибательные контрактуры в локтевых суставах и разгибателях кистей, ретракции пяточных сухожилий. Затем возникает слабость и атрофия двуглавых и трехглавых мышц плеча, позже — дельтовидных мышц и других мышц плечевого пояса. В некоторых случаях в качестве первого симптома отмечают ходьбу на пальцах и наружных краях стоп, которая развивается приблизительно в 5-летнем возрасте. До этого момента двигательное развитие детей обычно адекватное. Мышечная слабость возникает незаметно и медленно прогрессирует [Мальмберг С.А., Петрухин А. С., Широкова В.И., 2000]. Примерно в 20-летнем возрасте наступает относительная стабилизация. Возможность ходьбы и подъема по лестнице сохраняется. Лицевая мускулатура остается интактной. Обычно имеется проксимальная слабость (лопаточно-плечевая) в руках и дистальная (перонеальная) в ногах. Приемы Говерса могут отсутствовать, сухожильные рефлексы не вызываются. Псевдогипертрофия икроножных мышц не характерна. Часто обнаруживается укорочение заднешейных мышц, ведущее к недостаточной подвижности шейного отдела позвоночника. Иногда встречается сколиоз вследствие уплотнения и, возможно, ретракции паравертебральных мышц, который с возрастом не нарастает. Характерен проксимальный тетрапарез [Карпович Е.И., Казакова Л.В., Колбасова Л.В. и др., 1998].
Частыми и прогностически важными признаками болезни являются нарушения сердечной проводимости и развивающаяся дилатационная или гипертрофическая кардиомиопатия. Последняя может осложняться развитием паралича предсердий вследствие фиброза импульсгенерирующих синусоатриальных клеток. В этих случаях показана имплантация искусственного водителя ритма. Синкопальные состояния и приступы брадикардии в некоторых случаях могут предшествовать появлению мышечной слабости, но чаще возникают на 3-м десятилетии жизни. Изменения в проводящей системе сердца далеко не всегда обнаруживают при стандартном ЭКГ-исследовании. Однако атриовентрикулярные блокады и периоды Венкебаха могут быть выявлены при 24-часовом холтеровском мониторировании. Аритмия, которую не удается устранить при имплантации искусственного водителя ритма, может привести к инсульту и смерти больного. Витальный прогноз при миодистрофии Дрейфуса – Хогана всецело зависит от степени поражения сердца [Темин П.А., Белозеров Ю.М., Никанорова М.Ю., Страхова О. С., 1998].
Юношеская псевдогипертрофия Мэбри
Первые симптомы появляются в пубертатном периоде (11—13 лет) в виде слабости в мышцах бедер и тазового пояса. Характерны выраженные псевдогипертрофии мышц, умеренный проксимальный тетрапарез. Сухожильные ретракции нетипичны. Интеллект сохранен, отсутствуют ретракции и контрактуры. Облигатным признаком является кардиомиопатия [Palmucci L., Doriguzzi C., Mongini T. et al., 2004].
Миодистрофия Роттауфа — Мортье
Начинается в возрасте 8—9 лет, отличается выраженным миосклерозом, характерной чертой болезни являются ранние, выраженные и быстропрогрессирующие сухожильные ретракции и контрактуры в локтевых, голеностопных суставах, ригидности позвоночника. Вначале мышечные атрофии развиваются в тазовом и плечевом поясах, проксимальных отделах конечностей и мышцах спины. Затем мышечные атрофии преобладают в лопаточно-плечевой области и в дистальных отделах конечностей. Из-за контрактур формируется ходьба на носках, а затем невозможность сгибания позвоночника вследствие фиброза мышц. Парезы мышц выражены умеренно и в основном затрагивают плечевой пояс и дистальные отделы ног. Псевдогипертрофии отсутствуют, интеллект сохранен. Течение заболевания медленное, больные длительно сохраняют подвижность и обслуживают себя. Характерна кардиомиопатия с нарушением проводящей системы сердца. К 35—40 годам может развиться полная атриовентрикулярная блокада, что определяет летальный исход. Содержание КФК значительно повышено и снижается в далеко зашедших стадиях процесса. Гетерозиготные носительницы здоровы, а уровень КФК у них нормальный. Клинические проявления близки миодистрофии Эмери—Дрейфуса, однако отмечается более диффузное распределение мышечных гипотрофии и большая скорость прогрессирования миодистрофического процесса [Яхно Н.Н., Штульмен Д.Р., Мельничук П.В., 2001].

Поясно-конечностная юношеская миодистрофия Эрба — Рота.

Первое сообщение о прогрессирующей мышечной дистрофии было опубликовано в России в 1895 г. врачом В.К. Ротом, который назвал заболевание мышечной сухоткой. Термин "конечностно-поясная мышечная дистрофия" употребляется для обозначения случаев проксимальной мышечной слабости, которая начинает развиваться на 2-м или 3-м десятилетии жизни, прогрессирует медленно и приводит к глубокой инвалидизации лишь через 15—20 лет. Представляет собой группу заболеваний, ключевым симптомом которых является слабость проксимальных мышц верхних и нижних конечностей. В некоторых случаях плечевой и тазовый пояса вовлекаются одновременно. Как правило, описывается наследственная аутосомно-рецессивная форма прогрессирующих мышечных дистрофий Эрба - Рота восходящего типа. Это заболевание наблюдается примерно в 30% случаев прогрессирующей мышечной дистрофии и представляет собой гетерогенную группу, объединенную по принципу локализации патологического процесса преимущественно в мышцах плечевого и тазового поясов. В тех случаях, когда начинается процесс атрофии мышц верхних конечностей и затем атрофируются мышцы тазового пояса, а через 2-4 года, как правило, развивается атрофия мышц нижних конечностей, констатируют нисходящий тип болезни. Имеются также спорадические случаи. Заболевание проявляется в возрасте 13—16 лет, однако первые его признаки могут наблюдаться в раннем детском возрасте (дистрофия Лейдена) [Мальмберг С.А. и соавт., 2001]. Характерными симптомами являются слабость и атрофия мышц тазового пояса и бедер, мышц живота и туловища, что проявляется гиперлордозом позвоночника, выпячиванием живота, утиной походкой, затруднением при переходе из горизонтального положения в вертикальное. Генерализация атрофий происходит по восходящему типу. Псевдогипертрофии икроножных мышц, ретракции и контрактуры выражены умеренно, интеллект сохранен. Больной стоит, несколько расставив ноги, чуть-чуть согнув их в коленях. Поясница сильно вогнута, живот выпячен, верхняя часть туловища откинута назад. При спокойно висящих верхних конечностях локти расположены позади туловища. Наибольшая выпуклость позвоночника находится на уровне 2-3 грудных позвонков, а наибольшая вогнутость соответствует верхним поясничным позвонкам. Лопатки немного приподняты, их внутренние края параллельны и сближены до расстояния 8 см, но значительно отстоят от грудной клетки. Плечи тонки, как у малого ребенка. Предплечья сравнительно с плечами кажутся нормальными. Ягодицы очень похудели. Бедра при сдвинутых ногах не касаются друг друга, они имеют цилиндрическую форму вследствие преобладающей атрофии приводящих мышц. Несмотря на значительное уменьшение объема многих мышц, нет ни одной парализованной. Чтобы переменить положение, например, лежачее на сидячее, больной должен повернуться спиной кверху, принять положение a la vache, опуститься ягодицами на пятки, затем разогнуть туловище, опираясь руками о постель, наконец, высвободить из под себя ноги. Чтобы встать с постели, больному приходится повернуться спиной кверху при помощи ряда окольных движений и ухваток, спустив потом одну за другой ноги на пол, а затем притупить к самому трудному маневру - разгибанию туловища. Для этого больной отыскивает руками более высокую точку опоры: стол, спинку кровати и т.п. и пользуется ею, чтобы поднять туловище на сколько можно, вслед за тем, отталкивая его рукой, а также брюшных мышц одной стороны, он достигает того, что туловище описывает дугу и перегибается на сторону. Когда туловище придет таким образом в одну фронтальную плоскость с нижними конечностями, уже небольшого напряжения мышц достаточно, чтобы отклонить его кзади. Спина принимает единственное положение, в котором больной может стоять без поддержки и ходить. Экспрессивность мутантных генов варьирует, что определяет существование тяжелых, легких и даже субклинических форм поясно-конечностной миодистрофии Эрба — Рота. Кардиомиопатия проявляется в поздних стадиях заболевания [Мальмберг С.А. и соавт., 2001]. Атрофия дыхательных мышц, деформация грудной клетки и позвоночника приводят к нарушению функции внешнего дыхания, легочно-сердечной недостаточности Смерть больных обычно наступает от легочных осложнения, в частности, бронхопневмонии [Тетенев Ф.Ф., Бодрова Т.Н., Емельянова Н.В., 2000].
Плече-лопаточно-лицевая миодистрофия Ландузи — Дежерина.
Тип наследования аутосомно-доминантный, имеются спорадические случаи. Обычно медленно прогрессирующее, умеренной тяжести заболевание. Заболевание начинается в возрасте 12—20 лет. Семейная отягощенность может быть не обнаружена, поскольку пораженные члены семьи зачастую не подозревают о своих собственных проблемах. Первоначально атрофии наблюдаются в плечевом поясе с последующим распространением на лицо, следствием чего являются амимия. Слабость мимической мускулатуры выражается в неспособности свистеть и потере выразительности лица. Типичны «полированный» лоб, лагофтальм, «поперечная» улыбка, толстые, иногда вывороченные губы (губы тапира). Тип течения болезни в большинстве случаев относительно благоприятный. Однако физические перегрузки, интенсивные спортивные занятия и нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни. Многие больные не становятся инвалидами и качество их жизни не ухудшается. Других больных приковывает к креслу-каталке в зрелом возрасте. Как правило, больные отмечают изменение своей мимики: их речь становится неразборчивой. На высоте заболевания грубо страдают круговые мышцы рта и глаза, большая грудная, передняя зубчатая и нижние отделы трапециевидной мышцы, широчайшая мышца спины, двуглавая и трехглавая мышцы плеча. Отмечаются характерные симптомы в виде поперечной улыбки (улыбки Джоконды), протрузии верхней губы (губы тапира). Грудная клетка уплощается в переднезаднем направлении, плечевые суставы ротируют внутрь, лопатки приобретают крыловидную форму при попытке поднять руки вверх, деформация грудной клетки и позвоночника, скошенные плечи, появление широкого межлопаточного промежутка, уплощение грудной клетки, сколиоз. Атрофии распространяются в нисходящем направлении, и в процесс вовлекаются мышцы ног (лопаточно-плечебедренный, лицелопаточно-плечеперонеальный. лицелопаточно-плечеягодично-бедренный, лицелопаточно-плече-ягодично-бедренно-перонеальный и другие варианты). В таких случаях слабость наиболее заметна в группе малоберцовых мышц по свисающей стопе, но может быть и в проксимальных отделах ног [Бадалян Л.О., 2008]. Генерализация патологического процесса продолжается 10—15 лет, он постепенно распространяется на мышцы тазового пояса, проксимальных и дистальных отделов ног. Свисание стоп и слабость ног может вызывать падения больного и прогрессирующее затруднение движений. В некоторых случаях развиваются также атрофии мышц бедер и голеней. Псевдогипертрофия икроножных, дельтовидных, лицевых мышц выражена умеренно. Рефлексы могут быть долгое время сохранены. Характерной клинической особенностью является асимметрия атрофии. Возможно некоторое обратное развитие симптомов. Могут наблюдаться псевдогипертрофии мышц. Контрактуры и ретракции выражены умеренно. Кардиомиопатия редка. Аномалии сосудов сетчатки, которые могут быть обнаружены у многих больных при использовании метода ангиоретинмографии, рассматриваются в качестве составляющей части фенотипических проявлений болезни. В большинстве случаев с тяжелыми глазными проявлениями находят телеангиэктазии, отек и отслойку сетчатки. Может наблюдаться также снижение слуха. При выявлении телеангиэктазии их ликвидируют с помощью коагуляции, что предотвращает развитие слепоты. Больные длительное время сохраняют трудоспособность. Псевдогипертрофии выражены в икроножных и дельтовидных мышцах. Мышечный тонус в ранних стадиях болезни снижен в проксимальных группах мышц. Глубокие рефлексы снижены преимущественно с двуглавой и трехглавой мышц плеча. Интенсивные физические нагрузки ведут к быстрому прогрессированию заболевания [Сухомясова А.Л., 2005].
Лопаточно-перонеальная дистрофия.
Болезнь наследуется по сцепленному с Х-хромосомой типу и может являться аллельным вариантом для миодистрофии Эмери—Дрейфуса. До 5-летнего возраста дети здоровы, однако затем начинается деградация психики, которая проявляется неспособностью к обучению и отставанием умственного развития. Вскоре возникают слабость и атрофии лопаточных или плечевых и малоберцовых мышц. Контрактуры и псевдогипертрофии мышц не развиваются. Симптомы кардиомиопатии наблюдаются в подростковом возрасте и определяют летальный исход. Клиническая картина такая же, как при плечелопаточно-лицевой дистрофии, но нет мышечной слабости лица, возможны явления кардиомиопатии. В большинстве случаев заболевание начинается в среднем возрасте и наследуется по аутосомно-доминантному типу, но может встречаться и форма болезни с ранним началом, другим механизмом генетической передачи (связанный с Х-хромосомой, рецессивный), с клиническими проявлениями суставных контрактур и кардиомиопатией (тип Эмери—Дрейфуса) [Palmucci L., Doriguzzi C., Mongini T. et al., 2004].
Дистальная миодистрофия Говерса

Относится к редким заболеваниям, характеризуется генетической гетерогенностью. Тип наследования аутосомно-доминантный, имеются спорадические случаи. Проявляется в возрасте 30—60 лет. Симптоматика поражения мышц дистальных отделов конечностей. Характерными симптомами являются слабость и атрофии мышц голени и стоп, снижение ахилловых и коленных рефлексов. Ведущими симптомами являются шлепающие стопы, слабость мышц — разгибателей кисти. Вначале слабость и атрофия имеют место в икроножных мышцах, передняя группа мышц голеней остается интактной. Генерализация процесса с развитием атрофий кистей и проксимальных отделов конечностей происходит в течение 5—10 лет. Ахилловы рефлексы отсутствуют, все остальные вызываются [Бадалян Л.О., 2008]. У больных, как правило, выражена кардиомиопатия, приводящая к летальному исходу. Течение: доброкачественное, медленно прогрессирующее [Piantadosi C., Nigro V., Servider S. et al., 1998].
Офтальмоплегическая и окулофарингеальная (глазо-глоточная) формы.
Впервые заболевание было описано von Graefe в 1868 г. как «прогрессирующая наружная офтальмоплегия», в 1915 г. — Тейлором как семейный случай комбинации птоза век и паралича глотательных мышц. Однако только в 1962 г. М. Victor связал «прогрессирующую наружную офтальмоплегию» с фарингеальной слабостью и дал заболеванию название «окулофарингеальная миодистрофия» [Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008].
Генетически обусловленные миодистрофии глазных мышц делятся на несколько форм:
I. Изолированная окулярная миодистрофия, начинающаяся в молодом возрасте и приводящая к полной наружной офтальмоплегии, обычно без явлений диплопии. В ряде случаев процесс распространяется на другие поперечно-полосатые мышцы. Повышается уровень креатинфосфокиназы, лактатдегидрогеназы и альдолазы в сыворотке крови. Аутосомно-доминантный тип наследования.
II. Поздняя окулярная миодистрофия наблюдается в виде следующих форм: окулофарингеальная - поражение глазодвигательных и глоточных мышц с нарушением глотания; окулофациальная форма (отличается от формы Ландузи-Дежерина только сохранностью функции круговой мышцы глаза); окулобрахиальная форма (сочетается с поражением мышц проксимальных отделов конечностей); окулокардиальная форма.
III. Окулярная миодистрофия, сочетающаяся с немышечными поражениями дегенеративного характера, проявляющаяся задержкой развития пирамидной системы, морфологической или функциональной недостаточностью половых желез, сердечной деятельности. Это так называемый синдром Кирнса- Сейра - к офтальмоплегии присоединяется поражение центральной и периферической нервной системы (окуло-краниосоматическое нервно-мышечное заболевание). Появляется у детей и подростков с низким ростом, недостаточностью умственного развития, атаксией, глухотой, пигментной ретинопатией, дефектами проводящей системы сердца. Передается по аутосомно-доминантному и аутосомно-рецессивному типу [Becher M. W., Morrison L., Davis L.E. et al., 2001].
Тип наследования аутосомно-доминантный, имеются спорадические случаи. Возраст начала заболевания может быть разным. Чаще начало заболевания в возрасте 50-60 лет с явлениями птоза, ограничения экстраокулярных движений, лицевой и крикофарингеальной мышечной слабости [Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008].
В клинической картине заболевания наблюдается прогрессирующая мышечная слабость и атрофия проксимальных отделов конечностей, расстройства глотания и фонации, птоз, слабость лицевой мускулатуры. Характерны медленно нарастающая слабость и атрофия глазодвигательных мышц, ограничение движений глазных яблок. Крикофарингеальная мышечная слабость ведет к ахалазии, дисфагии и аспирации. Так как нарушения движений глаз носят хронический характер, они редко ведут к диплопии.
Поражение глазных мышц может быть изолированным или сочетаться с атрофией мышц лица, глотки (что приводит к затруднению глотания), поражением мышц конечностей. Течение медленно прогрессирующее [Blumen S.C., Brais B., Korczyn A.D. et al., 1999].
Миодистрофия Бетлема.
Дебют: раннее детство. Заболевание начинается со слабости мышц тазового пояса, которая возникает в грудном или раннем детском возрасте. Лицевая мускулатура остается интактной. Часто симптомы болезни настолько стертые, что родственники остаются неосведомленными об имеющихся отклонениях. Слабость прогрессирует медленно и обычно не приводит к инвалидизации и не влияет на продолжительность жизни. Рано развиваются сгибательные контрактуры в локтевых, голеностопных и межфаланговых суставах (кроме больших пальцев). Деформаций позвоночника не наблюдается. Ретракция пяточных сухожилий является причиной ходьбы на пальцах. Сухожильные рефлексы нормальны или снижены. Кардиомиопатия нехарактерна. Течение: доброкачественное, стационарное [Piantadosi C., Nigro V., Servider S. et al., 1998].

6. Диагностика
Для диагностики ранних проявлений прогрессирующих мышечных дистрофий с учетом международных протоколов модифицирован диагностический и лечебный паттерн, адаптированный к практическому здравоохранению, который включает:
Клинический осмотр с использованием разработанной шкалы эффективности реабилитации детей с нейромышечной патологией;
электронейромиографию с применением стандартных накожных регистрирующих электродов, стимулирующих биполярных электродов; Игольчатая ЭМГ с применением концентрических игольчатых электродов, включая одноразовые -выявляет изменение потенциалов двигательных единиц по первично-мышечному типу и спонтанную активность в виде потенциалов фибрилляций и положительных острых волн [Иллариошкин С. Н., Иванова-Смоленская И.А., Маркова Е.Д.. 2002];
Генетический анализ - ДНК-диагностика
Повышение уровня КФК (креатинфосфокиназы) отмечается при быстропрогрессирующих формах до 10 000 и выше ммоль/л, при медленнотекущих КФК может быть в норме или слегка повышена в затруднительных случаях дифференциальной диагностики первичных и вторичных мышечных дистрофий — биопсию мышц;
мышечная биопсия – при несомненном диагнозе не является необходимым методом исследования
биохимическое исследование ферментов крови (АЛТ, АСТ, КФК, ЛДГ) на фотометре;
исследование иммунологического статуса (T3, T4, T8, CD4/CD8, CD25, IgA, IgМ, IgG) [Emery A.E.H., 1994];
Электрокардиографию, при необходимости холтеровский мониторинг ЭКГ;
ЭхоКГ;
спирографию;
консультацию ортопеда, педиатра, кардиолога, психиатра (по показаниям);
при затруднении постановки диагноза — МРТ головного, спинного мозга;
в целях диагностики нарушений церебрального и периферического кровообращения — УЗДГ, цветное дуплексное сканирование;
для исключения остеопороза — денситометрию костей.
Диагностика прогрессирующих мышечных дистрофий подтверждается исследованием биопотенциалов мышц и микроскопическим изучением взятой у больного мышечной ткани [Новиков П.В., О.В. Евграфов, 1999].
С помощью методов ДНК-диагностики можно также определить носительство поврежденного гена для женщин в случае Х-сцепленных заболеваний . Женщина-носительница, как правило, является клинически здоровой, однако ее сыновья с вероятностью 0,5 будут больны. Для женщин-носительниц необходимо рекомендовать пренатальную диагностику, в то время как женщины-неносительницы не нуждаются в дальнейшем генетическом консультировании. Для ДНК-диагностики в постнатальном периоде (у больных) обычно используются ядросодержащие клетки крови; для дородовой диагностики чаще всего используются клетки ворсин хориона, амниотической жидкости, кровь плода [Новиков П.В., О.В. Евграфов,1999].
Таким образом, ДНК-диагностика позволяет решить ряд проблем для семей с наследственными болезнями, многие из которых просто неразрешимы другими методами. Это определяет значительную роль ДНК-диагностики не только для генетического консультирования, но и для педиатрии в целом [Евграфов О.В., Макаров В.Б., 1991].
Диагностика дистрофии Дюшенна.
Мутационный анализ, который базируется на оценке полиморфизма длины рестрикционных фрагментов, в настоящее время является общепринятым для диагностики болезней Дюшенна и Беккера, выявления носительства гена и пренатальной диагностики. Анализ содержания дистрофина в мышцах с использованием иммуногистохимической реакции на дистрофии помогает отличить миодистрофию Дюшенна от формы Беккера и дает возможность прогнозировать тип клинического течения. У гетерозиготных носительниц примерно в 70 % случаев выявляются субклинические признаки патологии скелетных мышц: повышение КФК, первично-мышечные изменения на ЭМГ и при исследовании мышечных биоптатов [Гехт Б.М., Касаткина Л.Ф., Самойлов М.И., Санадзе А.Г., 1997]. Изредка у носительниц отмечается некоторое уплотнение и увеличение объема икроножных мышц, повышенная утомляемость при физической нагрузке [Шишкин С.С., Ковалев Л.И., 1998].
При наличии клинического фенотипа миодистрофии Дюшенна у девочек следует в первую очередь исключить наличие Х-аутосомных транслокаций или других хромосомных аберраций с заинтересованностью локуса хромосомы 21, феномена лайонизации (патологической инактивации нормального аллеля гена в Х-хромосоме). Кроме того, требуется исключить "чистые" и мозаичные варианты синдрома Шерешевского—Тернера (Х-моносомии) и синдрома Морриса (XY). С этой целью проводят цитогенетическое исследование кариотипа. Дифференцируют миодистрофии Дюшенна и Беккера от врожденной дисплазии тазобедренных суставов, витамин D-резистентного рахита, проксимальных типов спинальных амиотрофий, полимиозита и дерматомиозита, метаболических и эндокринных миопатий [Piantadosi C., Nigro V., Servider S. et al., 1998].
Уже в ранних стадиях заболевания обнаруживают креатинурию, гипераминоацидурию, повышение альдолаз, трансаминаз (особенно аланиновой) и специфического фермента мышечной ткани — креатинфосфокиназы. Нарушения всех видов обмена веществ (углеводного, жирового, белкового), гипераминоацидурия, гиперферментурия, пентозурия, креатинурия могут наблюдаться и при других формах нервно-мышечных заболеваний. Однако при миодистрофии Дюшенна биохимические изменения выражены в большей степени, что является дополнительным критерием при оценке тяжести заболевания. Эти изменения служат биохимическими маркерами при выявлении гетерозиготных носителей. У матери — носительницы мутантного гена обнаруживают малые признаки болезни: уплотнение икроножных мышц, мышечную слабость при физической нагрузке, миодистрофические изменения при электромиографии, гиперферментемию. Наряду с классическим рецессивным типом наследования встречаются отклонения в типе наследования, например, существуют семьи, где все мальчики страдают этим заболеванием, а также семьи, где заболевание встречается у девочек [Piccolo F., Roberds S.L., Jeanpierre M. et al., 1995].
Значительное повышение (в 20-100 раз) мышечных ферментов (КФК, альдолаза), миопатическая кривая на ЭМГ [Гехт Б.М., Касаткина Л.Ф., Самойлов М.И., Санадзе А.Г., 1997]; в биоптатах — наличие некротизированных мышечных волокон с регенерацией, фагоцитозом и жировым перерождением мышечной ткани. Диагноз может быть поставлен точно при обнаружении дистрофина в мышечной ткани методом вестерн-блоттинга и (или) иммунохимической метки. Мутации в дистрофин-гене могут быть доказаны примерно у двух третей больных с помощью исследования кДНК. Изменения ЭКГ (увеличенный комплекс RS в отведении V,, глубокий Q в грудных отведениях) свидетельствуют о наличии кардиомиопатии [Карпищенко А.И., 1999].
Определение носителъства. Сывороточная КФК повышена у 50% женщин-носителей. Хотя ген и его производное (дистрофии) еще не идентифицированы, в практике можно использовать пробы на кДНК для определения носительства и пре-натальной диагностики [Piccolo F., Roberds S.L., Jeanpierre M. et al., 1995].
Ранняя диагностика. У больных имеется нестабильный участок ДНК с повышенным количеством CTG-триплетных повторов в хромосомном локусе 19ql3.3. Молекулярно-генетические исследования способствуют раннему выявлению и пре-натальной диагностике [Евграфов О.В., Макаров В.Б., 1991; Новиков П.В., О.В. Евграфов, 1999].
Диагностика дистрофии Беккера.
Диагноз ставится на основании генеалогического анализа (рецессивный сцепленный с X-хромосомой тип наследования), особенностей клиники (начало болезни в 10-15 лет, атрофии в проксимальных группах мышц, медленное, в течение 10-20 лет, распространение атрофии в восходящем направлении, массивные псевдогипертрофии икроножных мышц, умеренные соматические расстройства, медленное течение), данных биохимических исследований, электронейромиографии и биопсии мышц, выявляющих первично-мышечный тип изменений. Уровень КФК нормален или слегка повышен, характерные признаки миотонии и миопатии на ЭМГ, типичные признаки повреждения волокон в мышечных биоптатах. Биопсия мышц - некроз отдельных волокон, перерождение мышечных волокон. В крови повышена активность креатинфосфокиназы, альдолазы, лактатдегидрогеназы. ЭМГ - признаки миопатии. На ЭКГ - признаки поражения миокарда и нарушения проводимости [Карпищенко А.И., 1999]. Осложнения со стороны сердца, включая полную сердечную блокаду, представляют серьезную угрозу жизни больного. Следует тщательно контролировать дыхательную функцию, так как хроническая гипоксия может вести к развитию легочного сердца [Muntoni F., Mateddu A., Marchei F. et al., 1993].
Дифференцировать болезнь следует с прогрессирующими мышечными дистрофиями Дюшенна, Эрба-Рота, спинальной амиотрофией Кугельберга-Веландер [Comi G.P. et al, 1992].
При поражении сердечно-сосудистой системы диагноз может быть поставлен на основании инструментальных методов исследования, в то время как клинические симптомы могут либо отсутствовать, либо быть весьма ограниченными, особенно у пациентов на инвалидных колясках [Зенков Л. Р., Ронкин M. А.. 1991]. Так отеки и утомляемость, характерные для пациентов с манифестной сердечной недостаточностью, не выявляются у больных с латентной сердечной недостаточностью; также абсолютно не характерна для них и никтурия, весьма частая у пациентов с манифестной сердечной недостаточностью; застойная гепатомегалия у больных с манифестной сердечной недостаточностью обычно вызывает ощущение тяжести или болезненность в области правого подреберья, у пациентов с латентной сердечной недостаточностью может наблюдаться увеличение печени, однако ощущения тяжести или болезненности не наблюдается. Наиболее часто у больных с латентной сердечной недостаточностью наблюдается редкий кашель и повышенное потоотделение [Nigro V., de Sa Moreira E., Piluso G. et al., 1996]. Все остальные изменения можно выявить только с помощью инструментальных методов исследования [Зенков Л. Р., Ронкин M. А.. 1991].
Для диагностики поражения сердца при мышечной дистрофии Дюшенна наиболее информативными методами обследования являются: электрокардиография, электрокардиографическое мониторирование ритма сердца в течение 24 ч, эхокардиографическое и допплер-эхокардиографическое исследования, иммуно-гистохимический анализ распределения дистрофина в миокарде при эндомиокардиальной биопсии, исследование биоптата скелетной мышцы. По показаниям может быть произведена протонно–эмиссионная компьютерная томография. Последнее исследование может проводиться с целью оценки дефицита перфузии миокарда [Зенков Л. Р., Ронкин M. А.. 1991].
Для выявления пациентов с латентной формой сердечной недостаточности у пациентов с прогрессирующей мышечной дистрофией Дюшенна (Бекера) необходимо раз в 3 месяца проводить электрокардиографическое и эхокардиографическое исследования, оценку функции внешнего дыхания, и раз в 6 месяцев – цветное эхо– допплерографическое исследование [Nigro V., de Sa Moreira E., Piluso G. et al., 1996].
На электрокадиограммах у больных с мышечной дистрофией Дюшенна чаще всего выявляется глубокий зубец Q в отведениях II-III, aVF и V-6, а также высокий зубец R в отведении V-1, что свидетельствует о поражении миокарда в области задненижней и латеральной стенки левого желудочка [Saito K.et al, 2002]. В общей сложности ЭКГ нарушения диагностируются приблизительно у 54% больных (36% пациентов имеют нарушения сердечного ритма, 27% – признаки гипертрофии левого желудочка, 5% – признаки ишемии миокарда) [Adzija D et al.,1994].
Проведение суточного электрокардиографического монитори- рования позволяет с высокой достоверностью выявить наличие явных и скрытых нарушений ритма сердца у больных с мышечной дистрофией Дюшенна, а также оценить частоту встречаемости аритмий в данной нозологической группе. Различные типы нарушений сердечного ритма наблюдаются у 63,8% больных. Желудочковые эктопии обнаруживаются у 30% пациентов, при этом частота выявления желудочковых экстрасистол напрямую коррелирует с тяжестью клинических проявлений поражения скелетных мышц. По прошествии 5 лет частота выявления желудочковых аритмий в данной группе больных возросла до 74,5%, а смертность среди пациентов составила около 56% [Зенков Л. Р., Ронкин M. А.. 1991].
При более детальном изучении характера аритмий у больных с мышечной дистрофией Дюшенна оказалось, что простые желудочковые эктопии выявляются у 33% пациентов, а более комплексные (3, 4А, 4Б градаций по Лауну) – у 27% пациентов. При этом среди пациентов, умерших в течение 3 лет после начала наблюдений, желудочковые экстрасистолы были диагностированы у 40% больных, а среди выживших – только у 10%. Среди внезапно умерших пациентов комплексные желудочковые аритмии выявлялись значительно чаще у 66% [Chenard A.A. et al.,1993].
Прогностически значимые аритмии у больных c мышечной дистрофией Дюшенна могут сочетаться с асимптоматической дисфункцией левого желудочка и аномалиями подвижности сердечной стенки, при отсутствии клиники застойной сердечной недостаточности. Например, в исследованиях Chenard A.A. et al. (1993), у всех пациентов с выявленными желудочковыми аритмиями наблюдались патологические значения отношения времени систолических интервалов (отношение периода предызгнания к времени изгнания левого желудочка) – больше 0,48, а также имелись области дискинезии и акинезии сердечной мышцы. Исходя из вышесказанного, больным с мышечной дистрофией Дюшенна, у которых имеются нарушения ритма, необходимо проводить допплер- эхокардиографическое исследование [Sharma K.R., Mynhies M.A., Robert Y., Miller R.Y., 1993].
Данные допплер-эхокардиографического исследования у больных с мышечной дистрофией Дюшенна показывают, что систолическая дисфункция у данной категории больных впервые появляется в первой декаде жизни. Случаи возникновения патологии после 10 лет крайне редки, а после 14 лет манифестация эхокардиографических признаков заболевания практически не встречается [Карпищенко А.И., 1999]. В отличие от пациентов с ишемическими или гипертоническими заболеваниями, у пациентов с миопатией Дюшенна диастолическая дисфункция не предшествует и не сопутствует систолической, а также не происходит компенсации левожелудочковой дисфункции за счет левого предсердия. Данная категория больных, как правило, имеет предоминантную систолическую дисфункцию [Comi G.P. et al, 1992].
У больных с развернутой картиной эхокардиографических нарушений наиболее часто выявляются гипертрофическая кардиомиопатия (около 55% случаев), дилатационная кардиомиопатия (примерно 25% случаев), а также, значительно реже, встречаются такие нарушения, как дефект межпредсердной перегородки, пролапс митраиссльного клапана и миксома левого желудочка [Adzija D et al.,1994].
Выявить поражение миокарда на ранних стадиях и исследовать сердечно-легочную функцию у детей с мышечной дистрофией Дюшенна позволяет проведение протонно-эмиссионной компьютерной томографии. При этом дефицит перфузии наблюдается у 96% пациентов и дебютирует уже к 6-летнему возрасту. Дефект перфузии распространяется чаще всего от латеральной стенки левого желудочка до передней стенки и/или межжелудочковой перегородки. Худший прогноз в отношении риска возникновения сердечно- легочной недостаточности в ближайшее от проведения исследования время имеют пациенты с дефицитом перфузии более 10% и в возрасте моложе 15 лет [Sharma K.R., Mynhies M.A., Robert Y., Miller R.Y., 1993].
Причиной смерти при прогрессирующей мышечной дистрофии Дюшенна чаще всего является развитие дыхательной недостаточности вследствие торакодиафрагмальных и респираторных нарушений [Saito K.et al., 2002]
При гистологических исследованиях сердечной мышцы выявляется атрофия мышечных волокон, замещение их интерстициальным фиброзом, умеренная жировая инфильтрация и артериопатия мелких интрамуральных коронарных сосудов [Porter J.D., 1998].
Диагностика плечелопаточно-лицевой дистрофии.
Уровень КФК нормален или слегка повышен, смешанные признаки миопатии-невропатии на ЭМГ и в биоптатах мышц. У больных определяются мутации в хромосоме 4q35. Специфическое для этого локуса генетическое исследование проводят для выявления носительства и в пренатальной диагностике. Ортопедические средства и другие стабилизационные меры могут быть полезны для отдельных больных [Rando T.A., Dziesietnik M.H., Dhawan J. et al., 1997].
Диагностика юношеская псевдогипертрофии Мэбри
Активность КФК повышена. При биопсии мышц - имеются атрофические изменения мышечных волокон, резко выражен липоматоз. Миодистрофию Мэбри дифференцируют с миодистрофией Беккера, тазово-бедренной миодистрофией Лейдена—Мебиуса [Яхно Н.Н., Штульмен Д.Р., Мельничук П.В., 2001].
Диагностика офтальмоплегической формы дистрофии
Молекулярно-генетический анализ
Проводят амплификацию фрагмента ДНК, содержащего исследуемые области тринуклеотидных GCG-повторов в 1-м экзоне гена PABPN1 [Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008; Mirabella M., Silvester G., Rosa G. et al., 2000].
Прямая ДНК-диагностика
С целью быстрой молекулярно-генетической диагностики офтальмоплегической формы дистрофии был внедрен способ прямой ДНК-диагностики с помощью амплификации тринуклеотидного участка гена методом ПЦР и электрофореза в 8% полиакриламидном геле. Подтверждением клинического диагноза офтальмоплегической дистрофии служит обнаружение у больного на электрофореграмме двух различных фрагментов ДНК, один из которых имеет нормальный размер (6 GCG-копий), другой — патологически удлинен и соответствует экспансии GCG-повторов более 8 копий. Обнаружение у клинически здорового человека удлиненного аллеля с более 8 GCG-повторами предполагает пресимптоматическое носительство мутантного гена окулофарингеальной миодистрофии [Евграфов О.В., Макаров В.Б., 1991; Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев , 2008; Mirabella M., Silvester G., Rosa G. et al., 2000].
Диагностика конечно-поясной формы Эрба-Рота.
При биохимическом исследовании обнаруживают гиперферментурию, гипераминоацидурию, креатинурию, пентодурию и др., но выраженные в меньшей степени, чем у больных миодистрофией Дюшенна. Уровень креатинфосфокиназы в сыворотке крови таких больных значительно повышен на ранних стадиях, но по мере прогрессирования болезни концентрация снижается до нормальных величин. Содержание КФК повышено, однако не столь резко как при Х-сцепленных псевдогипертрофических формах. В биоптатах скелетных мышц выявляются некротические и регенераторные изменения мышечной ткани, разные размеры мышечных волокон, уменьшение количества мышечных ядер, разрастание соединительной ткани [Бадалян Л.О. , 2008; Вельтищев Ю.Е. и соавт., 1998; Гаусманова - Петрусевич И., 2001; Гехт Б.М., Касаткина Л.Ф., Самойлов М.И., Санадзе А.Г., 1997; Иллариошкин С. Н., Иванова-Смоленская И.А., Маркова Е.Д.. 2002 ]. При ЭМГ-исследовании отмечается первично-мышечный характер поражения. С помощью электромиографии выявляют снижение амплитуды, полифазность биопотенциалов [Зенков Л. Р., Ронкин M. А.. 1991]. Клиническое обследование членов семьи больного, а также электрофизиологическое и биохимическое исследования позволяют выявить малые признаки болезни [Sharma K.R., Mynhies M.A., Robert Y., Miller R.Y., 1993].
Наиболее часто проблемы дифференциальной диагностики возникают при разграничении миодистрофии Эрба-Рота и миодистрофии Беккера. В самом деле, у многих больных мужского пола, имеющих дистрофию с конечностно-поясными проявлениями, в результате определения сниженного содержания дистрофина устанавливают диагноз болезни Беккера. Дифференциация между этими формами дистрофий очень важна для обеспечения корректного медико-генетического консультирования. Конечно-поясную форму Эрба-Рота также необходимо отличать от ювенильной спинальной амиотрофии, миопатии с накоплением гликогена, эндокринных, токсических, лекарственных, карциноматозных миопатий, полимиозита и миозита с включениями телец [Мальмберг С.А. и соавт., 2001].
Диагностика миодистрофии Дрейфуса-Хогана
Pанняя диагностика миодистрофии Дрейфуса – Хогана важна не только для медико-генетического консультирования, но принципиально значима именно в плане терапии. Активность КФК повышена умеренно. У гетерозиготных носительниц повышение уровня КФК обычно незначительное [Тверская С.М., Руденская Г.Е., Чухрова А.Л., Поляков А.В. 2003; Мальмберг С.А., Петрухин А. С., Широкова В.И. , 2000 ]. В пользу миодистрофии Дрейфуса- Хогана свидетельствует отсутствие РИФ на эмерин с 12 моноклональными антителами при биомикроскопии лейкоцитов, мышечных и кожных биоптатов. У гетерозиготных носительниц уровень экспрессии эмерина в лейкоцитах по данным иммуногистохимического анализа также снижен. Для болезни характерны сочетанные ЭМГ-признаки первичномышечного и неврогенного поражения с большой представленностью спонтанной активности [Зенков Л. Р., Ронкин M. А.. 1991]. Дифференциальную диагностику проводят с аутосомно-доминантной формой миодистрофии с контрактурами (тип Гауптмана—Тангаузера), синдром ригидного позвоночника, лопаточно-плечевым синдромом с деменцией, скапулоперонеальной спинальной амиотрофией Старка—Кайзера [Яхно Н.Н., Штульмен Д.Р., Мельничук П.В., 2001].
Диагностика лопаточно-перонеальной миодистрофии
На ЭМГ выявляются смешанные миопатические и неврогенные изменения [Гехт Б.М., Касаткина Л.Ф., Самойлов М.И., Санадзе А.Г., 1997; Бадалян Л.О., Скворцов И.А., 1986]. При мышечной биопсии обнаруживают чрезмерное увеличение числа ядер и расщепление мышечных волокон. Уровень КФК повышен [Яхно Н.Н., Штульмен Д.Р., Мельничук П.В., 2001].
Диагностика плече-лопаточно-лицевой миодистрофии Ландузи — Дежерина
Уровень КФК может повышаться в 5 раз, однако в некоторых случаях содержание фермента нормально. На ЭМГ регистрируются как миопатические ДЕ, так и денервационные потенциалы [Гехт Б.М., Касаткина Л.Ф., Самойлов М.И., Санадзе А.Г., 1997]. Во многих мышцах конечностей гистологические изменения минимальные; наибольшее число патологических находок выявляется в надлопаточных мышцах, где обнаруживаются признаки прогрессирующей дегенерации и небольшой краевой денервации. Могут присутствовать воспалительные клетки. Дифференциальную диагностику лицелопаточно-плечевой миодистрофии проводят со спинальной амиотрофией с аналогичными клиническими прявлениями [Яхно Н.Н., Штульмен Д.Р., Мельничук П.В., 2001].
Диагностика миодистрофии Бетлема
Уровень КФК нормальный или слегка повышен. ЭМГ обычно изменена по миопатическому типу [Гехт Б.М., Касаткина Л.Ф., Самойлов М.И., Санадзе А.Г., 1997]. При биопсии обнаруживают признаки миопатии [Porter J.D., 1998].
7. Лечение
Радикального лечения не существует
Используется поддерживающая терапия: креатина моногидрат, антиоксиданты, таурин
При прогрессирующей мышечной дистрофии Дюшенна применяется преднизолон в дозе 1 мг/кг веса в день по чрездневной схеме
Проводятся попытки генной терапии, терапии стволовыми клетками [Евтушенко С.К., Евтушенко И.С., 2008].
На сегодняшний день миодистрофия Дюшенна неизлечима. Наиболее перспективным направлением поиска эффективной терапии считается разработка методов, позволяющих повысить экспрессию дистрофина в скелетных мышцах больных. По общему мнению ученых, именно с генной терапией дистрофинопатий связаны наиболее серьезные надежды добиться уже в обозримом будущем первых реальных результатов в борьбе с этим тяжелейшим заболеванием. Попытки прямого введения в мышцу миобластов (дистрофинпозитивных клеток-предшественников) привели лишь к минимальной и кратковременной экспрессии дистрофина, главным образом вследствие ограниченной миграции вводимых клеток из места инъекции и плохой приживаемости донорских миобластов [Евтушенко С.К., Евтушенко И.С., 2008].
Лечение направлено на поддержание физической активности пациента и улучшения качества его жизни. Использование протезов позволяет больным двигаться и замедляет формирование сколиоза. Разрабатывается генная терапия (гены дистрофина и утрофина) [Rando T.A., Dziesietnik M.H., Dhawan J. et al., 1997].
Лечение заболеваний этой группы направлено на улучшение белкового и энергетического обмена в мышцах, нормализацию витаминного баланса в организме, стимуляцию нервно-мышечной передачи, усиление капиллярного кровотока и др. С этой целью применяются медикаментозные препараты и различные физиотерапевтические методы лечения [под ред. В.К. Видерхольда,2004].
Лечение, которое проводится больным, является комплексным и включает патогенетическую, специальную медикаментозную терапию с учетом степени тяжести заболевания (легкая, средняя, тяжелая), стадии (компенсации, субкомпенсации, декомпенсации), физиотерапевтические процедуры, синглетно-кислородную терапию, лечебную физкультуру (дыхательная гимнастика, стренч-гимнастика), специальную диету, электроакупунктуру, щадящий массаж функционально сохранных мышц [Лобзин В.С. и др., 2002].
Сбалансированное лечебное питание (продукты, содержащие белок, полиненасыщенные жирные кислоты, витамины, микроэлементы): овощи, творог, рыба, печень, соевое мясо. Весной и осенью — курсовой прием поливитаминных препаратов и микроэлементов (активал, мультитабс, биовиталь, мильгамма, нейровитан, неуробекс) [под ред. В.К. Видерхольда, 2004].
Массаж при нейромышечных заболеваниях существенно отличается от стандартных методик его проведения. Сила воздействия минимальна, акцент на улучшение трофики кожных покровов и сохранных мышц с применением актовегиновой мази, бальзама «Живокост», щадящее растягивание укороченных сухожилий с применением препаратов хондроксид, траумель С, поглаживание суставов, паравертебрально-точечный гармонизирующий массаж. Длительность сеанса — до 10 мин. Курс № 10. При наличии симптоматики слабости дыхательной мускулатуры выполняется массаж грудной клетки для облегчения дыхательных движений [Евтушенко С.К., Евтушенко И.С., 2008].
Примерная схема массажа при прогрессирующей мышечной дистрофии Дюшенна: сеанс начинается с поглаживания спины, грудной клетки и конечностей с применением трофических мазей и заканчивается им, далее проводится гармонизирующий точечный массаж паравертебрально, после чего инструктор легкими движениями пальцев по ходу волокон мышц дистальных отделов конечностей прорабатывает пучки мышц, сухожилий, далее переходит на аккуратное растягивание укороченных сухожилий [Лобзин В.С. и др., 2002].
Дозированная лечебная физкультура с элементами stretch-гимнастики, направленная на поддержание и максимальное сохранение функциональной способности невовлеченных в патологический процесс мышц в каждом конкретном случае с учетом формы нейромышечного заболевания [Евтушенко С.К., Евтушенко И.С., 2008].
Коррекция ортопедических проявлений составляет весомую часть в реабилитации больных с нейромышечной патологией [Евтушенко С.К., Евтушенко И.С., 2008].
Наиболее часто в клинике преобладают:
— деформация туловища, позвоночника (нейромышечный сколиоз), конечностей;
— врожденный вывих и подвывих тазобедренных суставов, контрактуры суставов, мышц;
— деформация стоп (эквиноварусные стопы, косолапость, плоскостопие, деформация стоп по типу Фридрейховских, гипермобильность суставов, нестабильность надколенника, сухожильные ретракции, псевдогипертрофии мышц) [Лобзин В.С. и др., 2002].
До 40 % детей, у которых первично выявлена нейромышечная патология и в клинике которых имеет место симптоматика поражения опорно-двигательного аппарата, длительно наблюдаются и лечатся у ортопедов. Поступают такие дети в стадии выраженных клинических симптомов болезни, когда существенно повлиять на течение патологического процесса практически невозможно [под ред. В.К. Видерхольда, 2004].
Лечение ортопедических проявлений у больных с миодистрофией является сложной и до конца не решенной проблемой, анализ литературы по этому поводу свидетельствует о существовании разных точек зрения. При начальных проявлениях контрактур, ретракции сухожилий используются щадящий массаж с применением трофических мазей, фармакопунктуры с цель Т, траумель С, направленные на улучшение питания сустава, специально разработанные методики ЛФК, фиксацию конечности в положении достигнутой коррекции контрактуры суставов, шины, валики для профилактики контрактур, фиксацию конечностей в физиологическом положении на ночь с использованием туторов; с целью адаптации передвижения с оптимальной коррекцией деформаций используются стельки, ортопедическая обувь, надколенники. При доброкачественных формах миодистрофий в стадии компенсации возможно проведение оперативного вмешательства, направленного на предупреждение и избавление контрактур, сухожильных ретракций, коррекцию деформаций [Лобзин В.С. и др., 2002].
В целях укрепления мышечного корсета спины используется фармакопунктура паравертебрально с применением нейромидина, церебролизина, актовегина, кортексина, цианокобаламина. В стадии субкомпенсации рекомендуется 1–2-часовое ношение реклинаторов, корсетов в моменты наибольшей нагрузки на позвоночный столб (сидение, ходьба и др.), несмотря на категорическое несогласие с этим ортопедов. В стадии декомпенсации, тяжелой степени с целью адаптации передвижения с оптимальной коррекцией деформаций используются стельки, ортопедическая обувь, наколенники, рекомендуется практически постоянное ношение корсетов, так как в данной стадии заболевания происходят выраженная атония и гипотрофия мышечного корсета, влекущие за собой резкую деформацию позвоночного столба, приводящую к вторичной висцеропатии, ухудшению работы сердца, легких, влекущих за собой еще большую декомпенсацию патологического процесса. При доброкачественных формах нейромышечных заболеваний в стадии компенсации возможно проведение оперативных вмешательств, направленных на предупреждение контрактур, сухожильных ретракций и избавление от них, коррекцию деформаций [Евтушенко С.К., Евтушенко И.С., 2008].
Когнитивные нарушения, в частности, при прогрессирующей мышечной дистрофии Дюшенна имеют место в 40 % случаев. Наряду с традиционными ноотропными препаратами (церебролизин, тиоцетам, пирацетам, луцетам и др.) мы предпочитаем назначение и таких ноотропных препаратов, как семакс 0,1% по 3–4 капли в носовые ходы 2 раза в день 10 дней, чередуя с приемом когитума по 1/2–1 ампулы внутрь 2 раза в день 10 дней, курсами цераксон по 2 мл 2–3 раза в день — 45 дней [под ред. В.К. Видерхольда, 2004].
Проблема поражения сердечно-сосудистой системы при нейромышечных заболеваниях актуальна, поскольку летальный исход большинства из них обусловлен именно вовлечением сердца в патологический процесс, так как сердечная мышца поражается первично вследствие дефицита того же структурного компонента, которого нет и в скелетной мышце. Сердце — непрерывно и интенсивно работающий орган, и нередко дебют миопатии начинается именно с кардиологических проявлений, в то время как симптомы миопатического процесса могут быть маскированными [Страхова О.С., Белозерова Ю.М., Темин П.А., 1999].
Согласно мировым стандартам эхокардиографического исследования, выделяют 6 клинико-патогенетических стадий поражения сердечно-сосудистой системы при нейромышечных заболеваниях [Quinlivan-RM; Dubowitz-V, 1992].
1. Диастолическая дисфункция миокарда по рестриктивному типу на стадии начальных проявлений заболевания (уменьшение скорости потока крови на митральном клапане с уменьшением амплитуды предсердной волны, уменьшение диастолического диаметра и объема левого желудочка, умеренное снижение фракции выброса левого желудочка).
2. Миокардиальная стабилизация на стадии компенсации заболевания (нормализация допплерэхокардиографических показателей). На данных стадиях целесообразно применение кардиотрофической терапии перорально 2–3 курса в год (АТФ-лонг, рибоксин, милдронат, кардонат, элькар, калия оротат, магнерот).
3. Систолическая дисфункция миокарда на стадии субкомпенсации миопатического процесса (умеренное увеличение систолического диаметра и объема левого желудочка, умеренное уменьшение фракции выброса).
4. Миокардиальная псевдостабилизация на стадии декомпенсации-А миопатического процесса (нормализация допплерэхокардиографических показателей).
5. Гипокинезия левого желудочка на стадии декомпенсации-В (выраженное увеличение систолического диаметра и объема левого желудочка, значительное снижение фракции выброса).
6. Дилатация левого желудочка на стадии декомпенсации-С (значительное увеличение диастолического и систолического диаметра и объема левого желудочка, резкое снижение фракции выброса) [под ред. В.К. Видерхольда, 2004].
Применение расчетных эхокардиографических параметров позволяет более тонко разобраться в патогенезе заболевания, констатировать предпатологические изменения и заблаговременно начать лечение. Такая тактика позволяет продлить срок самостоятельной ходьбы больных, отсрочить время наступления манифестной сердечной недостаточности [под ред. В.К. Видерхольда, 2004].
На 3–4-й стадиях целесообразно применение 10% раствора карнитина хлорида (данный препарат обладает метаболическим, нейротрофическим, кардиотрофическим и антиоксидантным свойствами) c кокарбоксилазой, аскорбиновой кислотой в/в капельно на фоне в/м введения пиридоксина гидрохлорида и перорального приема метионина. № 10, 3–4 курса в год. Амбулаторно дети принимают кардонат по 1 капсуле 2–3 раза в день — 45 дней [под ред. В.К. Видерхольда, 2004].
На 4–6-й стадии используется неотон (фосфокреатинин, «супер-АТФ») в/в капельно № 5, 3–4 курса в год [под ред. В.К. Видерхольда, 2004].
При анализе результатов реабилитационного лечения детей с нейромышечной патологией отмечена положительная динамика, которая выражается в том, что у больных со злокачественными формами миодистрофий (прогрессирующии мышечные дистрофии, форма Дюшенна, Эрба — Рота) по сравнению с детьми с аналогичными формами, на 2–3 года дольше сохраняется способность к самостоятельному передвижению, позднее формируются контрактуры, реже протекает ОРВИ, длительное время поддерживается насосная и сократительная способность миокарда. Дети с доброкачественными формами миодистрофий долгое время сохраняют вертикальную позу, социально адаптированы. Действительно, миодистрофии в настоящее время неизлечимы, но благодаря разработанным схемам медико-социальной реабилитации можно существенно улучшить качество жизни таких больных [Лобзин В.С. и др., 2002].
Лечебно-педагогические мероприятия в отношении больных с сохранным интеллектом должны быть направлены на повышение психического тонуса [Backman E., Henriksson K.G., 1998].
В большинстве случаев такие дети обучаются индивидуально на дому или в школах для детей с двигательными нарушениями. В задачу педагогов и родителей входит забота о социальной адаптации этих детей. Следует как можно раньше ориентировать ребенка относительно его будущей профессии; следует учитывать при этом его двигательные возможности. Больные со сниженным интеллектом обучаются педагогом-дефектологом на дому или в специальных учреждениях [Лобзин В.С. и др., 2002].
Основными задачами медикаментозной терапии сердечно-сосудистых осложнений являются компенсация энергетического дефицита мышечной ткани, улучшение тканевого метаболизма и периферического кровообращения. С этой целью применяются следующие группы препаратов [Quinlivan-RM; Dubowitz-V, 1992]:
Витамины (вит.В1 (тиамин), кокарбоксилаза, никотиновая кислота (вит.РР), кальция пантотенат, вит.В6 (пиридоксин), вит.В12 (цианкобаламин), кальция пангамат (вит.В15), витамин Е).
Аминокислоты (глутаминовая кислота, метионин).
Антихолинэстеразные препараты (галантамин, прозерин, оксазил).
Препараты, влияющие на тканевой метаболизм (калия оротат, АТФ, рибоксин, карнитин, убихинон , цитомак, лимантар).
Препараты, улучшающие периферическое кровообращение (продектин, трентал, теоникол (компламин), вазобрал).
Коррекция сердечной недостаточности осуществляется сердечными гликозидами, диуретиками, препаратами калия. Сердечные гликозиды пациентам с дилатационной кардиомиопатией и сопутствующей сердечной недостаточностью назначаются в минимальных терапевтических дозах, диуретики (чаще всего фуросемид) - до 25 мг в сутки (в зависимости от степени сердечной недостаточности) [Nigro V., de Sa Moreira E., Piluso G. et al., 1996]. Однако исключительно симптоматическая коррекция сердечной недостаточности сердечными гликозидами и диуретиками у пациентов с прогрессирующей мышечной дистрофией Дюшенна хотя и позволяет достаточно быстро улучшить состояние пациента, не способствует продлению жизни больных [Ishikawa Y, Bach JR, Sarma RJ et al., 1995].
Эффективность противоаритмических препаратов у больных с прогрессирующей мышечной дистрофии Дюшенна изучена недостаточно. Ishikawa Y, Bach JR, Sarma RJ et al., (1995) сообщают о возможном применении препаратов хинидинового ряда (хинидин и пр.), бета-адреноблокаторов (атенолол и др.), блокаторов кальциевых каналов (изоптин), лидокаина в лечении нарушений ритма сердца у пациентов с нейромышечными заболеваниями [Quinlivan-RM; Dubowitz-V, 1992].
До сих пор пациенты с мышечной дистрофией и сопутствующей кардиомиопатией весьма неохотно признаются годными к проведению трансплантации сердца. Это происходит из-за высокого периоперационного риска в связи со сниженной физической активностью пациентов и в связи с предполагаемым быстрым развитием кардиомиопатического процесса в пересаженном сердце. Однако такая точка зрения не вполне оправдана. В исследовании Rees W; Schuler S; Hummel M; Hetzer R. (1993) сообщается о 3 пациентах с миопатией Дюшенна, которым была проведена данная операция. Все пациенты находились в крайней стадии кардиомиопатического процесса. Средний возраст проведения операции составил 25 лет (от 9 до 45). Длительность послеоперационного наблюдения – от 10 мес. до 7 лет. Все пациенты имели неосложненный послеоперационный период, все получали иммуносупрессивную терапию азатиоприном, циклоспорином и стероидами. Ежегодные исследования состояния трансплантата с помощью рекатетеризации показывали нормальное функционирование левого желудочка (фракция выброса была в пределах нормы). Ни одного случая поражения коронарных артерий не отмечалось. Вплоть до настоящего времени ни у одного пациента не наблюдалось прогрессирования имевшейся до операции мышечной дистрофии [Quinlivan-RM; Dubowitz-V , 1998; Страхова О.С., Белозерова Ю.М., Темин П.А., 1999].
В последние годы наблюдается значительный прогресс в генной и клеточной терапии прогрессирующей мышечной дистрофии Дюшенна. Согласно современным представлениям генную терапию понимают как введение нуклеиновых кислот в клетку с целью воздействия на медицинский статус организма и/или лечения болезни [Kay M., Liu D, Hoogerbrugge P.M., 1997]. Существенно, что при этом сам ген уже воспринимается как новый фармацевтический препарат для лечения не одного, а многих заболеваний, не только моногенных (мутации в одном гене), но и полигенных, мультифакториальных (мутантные гены + неблагоприятные внешние факторы), инфекционных заболеваний и любых других патологических состояний. Большинство исследований по разработке подходов к генотерапии МДД проводятся на мышах со спонтанными или индуцированными мутациями в гене дистрофина. Первой биологической моделью заболевания была спонтанная мутация mdx мышей линии C57BL/10J. В настоящее время создан ряд линий мышей с мутациями в гене дистрофина. Наиболее близкими к человеку по тяжести и патогенезу заболевания являются мыши mdx52 с делецией 52 экзона, а также недавно полученные мыши mdx с направленной мутацией (knock-out) в гене утрофина - аутосомного аналога дистрофинового гена [Richard I., Brenguier L., Dincer P. et al., 1997].
По тяжести нарушений и клиническому проявлению наиболее близкой к человеку моделью миодистрофии Дюшенна является заболевание, индуцированное мутациями в гене дистрофина у собак породы золотой ретривер. Именно на них рекомендуется проводить завершающий этап предклинического тестирования генной терапии миодистрофии Дюшенна [Richard I., Brenguier L., Dincer P. et al., 1997].
В начале 90-х годов Питером Лоу было предложено лечение миодистрофии Дюшенна путем трансплантации аллогенных миобластов. Суть метода заключалась в заборе материала у здорового донора, выращивании миобластов в условиях клеточных культур с последующей их трансплантацией в мышцы больного. Стоимость операции оценивалась около 150 тыс. долларов. Исследования по проверке эффективности трансплантации аллогенных миобластов были проведены в 6 независимых исследовательских лабораториях. Согласно мнению ведущих авторитетов в области биологии мышц и миодистрофии Дюшенна в существующем на данный момент виде метод трансплантации миобластов (т.е. клеточной терапии по методу Питера Лоу) абсолютно неэффективен [Partridge T., Lu Q.L., Morris G., Hoffman E, 1998; Beauchamp J.R. Morgan J.E., Pagel C.N. et al, 1999]. В их экспериментах на мышах с использованием разнообразных клеточных маркеров и современных методов FISH анализа было показано что уже через 24 часа после трансплантации 90% гетерологичных миобластов отмирает или исчезает из места трансплантации, 5% находятся в покоящемся состоянии, 5% сливаются с пораженными мышечными волокнами, но только в половине из них (2.5%) наблюдается синтез дистрофина [Rees W; Schuler S; Hummel M; Hetzer R, 1993].
Значительно более перспективным направлением клеточной терапии миодистрофии Дюшенна представляется трансплантация аутологичных стволовых клеток, полученных из костного мозга или скелетных мышц. В исследованиях высокоочищенные стволовые клетки костного мозга здорового донора трансплантировали облученным mdx мышам. Было показано, что в результате трансплантации регенерируют не только клетки костного мозга реципиента, но значительная часть инъецированных клеток мигрирует и в скелетные мышцы, где сливаясь с миофибриллами восстанавливает синтез дистрофина. По прошествии 12 недель после трансплантации доля дистрофин положительных волокон увеличивалась с 1 до 10%. Таким образом, доказано, что в костном мозге присутствуют стволовые клетки не только всех форменных элементов крови, но и предшественники миобластов. В виду широкого распространения мультипатентных стволовых клеток после трансплантации, открывается возможность не только локального но и системного исправления дефекта в различных группах мышц при миодистрофии Дюшенна [Gussoni, E, Soneoka, Y., Strickland, C. D et al., 1999].
Генная терапия миодистрофии Дюшенна
Несмотря на очевидные успехи в исследованиях структуры гена дистрофина, его продуктов, и в выяснении биомеханизмов заболевания, реальных успехов в генотерапии МДД пока не достигнуто. Причиной этого являются, по-видимому, не только гигантские размеры гена и его мРНК, но и, главным образом, отсутствие эффективных средств доставки гена в мышцы [Горбунова В.Н., Баранов B.C., 1997].
Считается, что достижение терапевтического эффекта возможно при успешной трансфекции не менее 20%, а по последним данным даже 40% всех мышечных волокон не только скелетной мускулатуры, но так же мышц сердца и диафрагмы. При этом основными критериями эффективности трансфекции являются: появление дистрофин-положительных мышечных волокон, нормализация уровня биохимических маркеров миодистрофии Дюшенна, изменения физиологических параметров (силы мышц и др.) [Bashir R., Britton S., Strachan T. et al., 1998].
В активно разрабатываемых в настоящее время генотерапевтических подходах к лечению миодистрофии Дюшенна можно выделить несколько направлений [Шишкин С.С., Шаховская Н.И., Лунга И.Н. и др.]:
коррекция дефекта путем введения нормальных копий кДНК гена дистрофина в составе рекомбинантных вирусных частиц или посредством невирусных способов доставки;
коррекция мутаций на уровне геномной копии гена или на его первичном РНК- транскрипте;
активация в мышечных волокнах и клетках репрессированного в ходе онтогенеза аутосомного гомолога гена дистрофина - гена утрофина
Обоснована возможность введения необходимого компонента с-ДНК (минигена, кодирующего синтез участка дистрофина, на уровне которого произошла делеция) не только в скелетную мышцу, но и непосредственно в сердечную мышцу. Введение минигенов осуществляет конверсию дистрофиннегативных мышечных волокон в дистрофинпозитивные, что улучшает функциональные возможности мышечной ткани [Горбунова В.Н., Баранов B.C., 1997].
Лечение патогенетическое и симптоматическое. Комплексный курс лекарственной терапии включает назначение аминокислот (глутаминовой кислоты, метионина), пирацетама, витаминов (А, В, С, D, Е, кофермента О), сосудорасширяющих препаратов (ксантинали никошнат), никотиновой кислоты, средств, корригирующих энергетические процессы в мышцах (АТФ, АДФ), антихолинэстеразных препаратов (прозерина, сангвиритрина, пиридостигмина бромида), препаратов калия и кальция (калия хлорида, кальция глюконата), анаболических гормонов (неробола, ретаболила). Последние назначают лицам мужского пола в случаях мышечной кахексии для увеличения объема и массы мышц. ЛФК, массаж применяют строго дозированно по индивидуальным схемам. Благоприятный эффект оказывает электростимуляция мышц. Ортопедические мероприятия направлены на профилактику ретракции и контрактур. Рекомендуется преимущественно молочно-растительная диета (фрукты, овощи, продукты козьего молока, орехи, мед). Во всех случаях заболевания комплексное лечение способствует стабилизации процесса и улучшению самочувствия больных [http://doctor.ru/medinfo].
В связи с частыми ОРВИ необходимо назначение препаратов, влияющих непосредственно на иммунокомпентные клетки и центральные механизмы регуляции иммунитета, через которые оказывается вторичное иммуностимулирующее влияние на организм. К таким препаратам относятся: бронхо-мунал П, рибомунил, иммунал, ИРС. Бронхомунал П назначался на протяжении 10 дней с 20-дневным перерывом детям до 12 лет по 3,5 мг/сут, детям старше 12 лет — по 7,0 мг/сут [Лобзин В.С. и др., 2002].
Лечение мышечной дистрофии Дюшенна направлено на поддержании физической активности пациента и улучшение качества его жизни; как правило, быстро становится неэффективным. Физические упражнения выполняют систематически и по определённой схеме. Короткие перерывы показаны при возникновении болей в мышцах и мышечной усталости. Использование протезов позволяет больным двигаться и замедляет формирование сколиоза. Поддержание дыхания, ИВЛ во время сна для предотвращения синдрома ночной гиповентиляции. Экспериментальные методы, в особенности генная терапия (гены дистрофина и утрофина), чрезвычайно перспективны, хотя и не получили пока клинического распространения [Шишкин С.С., 1997].
Оперативное лечение. Ортопедическое вмешательство необходимо при наличии контрактур и фиксации суставов [Шишкин С.С., Шаховская Н.И., Лунга И.Н. и др., 2000].
Медикаментозное лечение прогрессирующих мышечных дистрофий.
Медикаментозное лечение назначается с учетом полученных результатов клинико-инструментального исследования и сопутствующей патологии (кардио-, пневмопатии). При доброкачественных формах прогрессирующей мышечной дистрофии (Говерса — Веландера, Давиденкова, Бетлема, Беккера, Эмери — Дрейфуса) в стадии стойкой компенсации, при легкой, легко-средней степени тяжести заболевания целесообразно назначение курсами препаратов «метаболического» действия, направленных на улучшение, поддержание обменных, «энергетических» процессов неповрежденных миоцитов, кардиомиоцитов. К таким препаратам относятся: АТФ-лонг, цитофлавин, кардонат, элькар, милдронат, магнерот, витамин Е, метионин. Назначается по 1–2 препарата курсами 2–3 раза в год [Шток В.Н., 2003].
Фенитоин, прокаинамид, хинин применяются в лечении миотонии, но требуется осторожность у больных с заболеваниями сердца (опасность ухудшения сердечной проводимости) [под ред. В.К. Видерхольда, 2004].
При наличии жалоб на боли в мышцах нижних конечностей, чувство «стягивания» мышц особенно хорошо зарекомендовал себя цитрулина малат. Препарат способствует «утилизации» молочной кислоты, одновременно обладая метаболическими свойствами. Данный препарат широко используют в профессиональном спорте при физическом перенапряжении, перед соревнованиями [под ред. В.К. Видерхольда, 2004].
При доброкачественных формах в стадии субкомпенсации, при средней степени тяжести заболевания, на ранних стадиях патологического процесса в стадии компенсации при злокачественных (быстро прогрессирующих) формах прогрессирующей мышечной дистрофии (Дюшенна, Эрба — Рота) назначается 10% раствор карнитина хлорида (данный препарат обладает метаболическим, нейротрофическим, кардиотрофическим и антиоксидантным свойствами) c кокарбоксилазой, аскорбиновой кислотой в/в капельно на фоне в/м введения пиридоксина гидрохлорида и перорального приема метионина. 3–4 курса № 10 в год с дальнейшим переходом на пероральный прием препарата кардонат в амбулаторных условиях длительностью до 2 мес. [Лобзин В. С., Сайкова Л. А., Шиман А. Г., 2000].
У детей до 5 лет предпочтение отдается жидким формам карнитинсодержащих препаратов ввиду удобства применения и лучшей переносимости [под ред. В.К. Видерхольда, 2004].
При этих же формах для фармакопунктур используются антигомотоксические препараты, препараты нейротрофического действия (церебрум композитум, траумель и др.) [Лобзин В.С. и др., 2002].
На стадии развернутой клинической картины миодистрофии Дюшенна назначается иммуноглобулин человека нормальный в дозе 5,0–7,0 мл/кг на инфузию . Весь объем иммуноглобулина растворяется в 4 раза изотоническим раствором и вводится со скоростью 15 капель в минуту. Количество инфузий — от 3 до 5. На фоне малых доз преднизолона по 5 мг 1 раз в день 3–6 мес. Иммуноглобулин продемонстрировал достаточно высокую эффективность, было отмечено снижение показателей КФК, ЛДГ, АЛТ, АСТ в среднем на 20 %, дети отмечали нарастание силы, переносимости физических нагрузок. Эффективность иммуноглобулинов при миодистрофии Дюшенна нами объясняется следующим образом [Penninger J.M., Neu N., Bachmaier К.,1996.]:
1. Не исключено, что разрушение дистрофинассоциированного комплекса белков может происходить не только в результате генетических факторов, но и вследствие вирусных инфекций. Это, в частности, доказано в отношении вируса Коксаки, способного разрушать дистрофин и ассоциированный с ним комплекс белков в кардиомиоцитах.
2. Распад мышечных волокон способствует развитию апоптоза, что подтверждают наши исследования по высокому уровню СD25 (маркер апоптоза). Некроз миоцитов, распад белков дистрофинассоциированного комплекса (белки цитоскелета, трансмембранные белки, периферические мембранные белки) с выбросом их фрагментов в кровь, приводящие к продукции циркулирующих иммунных комплексов и развитию аутоиммунизации. При злокачественных формах мышечных дистрофий в стадии субкомпенсации, начальных проявлениях декомпенсации, в стадии выраженных клинических проявлений при доброкачественных формах используем неотон (фосфокреатинин, «супер-АТФ») в/в капельно 1,0–4,0, № 5, 3–4 курса в год. Данный препарат используется у взрослых в комплексном лечении инфаркта миокарда, мы же стали его использовать для увеличения сократительной способности миокарда.
Кроме того, этим же детям назначаются подддерживающие дозы преднизолона 5–10 мг утром 1–3 месяца (курсами) [Лобзин В. С., Сайкова Л. А., Шиман А. Г., 2000].
Глюкокортикостероиды увеличивают мышечную силу у мальчиков, страдающих мышечной дистрофией Дюшенна, замедляя прогрессирование заболевания. При длительной стероидной терапии необходим тщательный контроль развития побочных эффектов, включающий наблюдение за массой тела, АД, состоянием слизистой оболочки ЖКТ и иммунной системы. Действие аптечных лекарств: стимулирующее на синтез белковых и жировых соединений, усиливающее микроциркуляцию в мышечной ткани, миотонизирующее, улучшающее пластику и трофику мышечной ткани, нормализующее нейро-гуморальные взаимоотношения, тормозящее катаболические процессы, нормализующее и стабилизирующее выработку в гипофизе адренокортикотропного гормона, повышающее выработку в надпочечниках глюкокортикоидов, блокирующее на выработку и освобождение стрессовых гормонов, иммуностимулирующее, десенсибилизирующее, стимулирующее эритропоэз, повышающее процессы тканевого дыхания, уменьшающее реактивность симпатики и парасимпатики вегетативной и центральной нервных систем, седатирующее, детоксикационное, общеукрепляющее, общеоздоравливающее [Шток В.Н., 2003].
Наблюдение. Ранняя диагностика поражения внутренних органов позволяет увеличить продолжительность жизни пациентов [под ред. В.К. Видерхольда, 2004].
8. Профилактика
При выявлении в семье больного прогрессирующей мышечной дистрофии рекомендуется обращение в медико-генетическую консультацию.
Большую роль играет профилактика [Лобзин В.С.и др.,2002]:
соблюдение рационального режима дня;
ограничение (лучше исключение) провоцирующих факторов, а также физических перенапряжений;
проведение щадящих реабилитационных мероприятий (занятие ЛФК, спортом необходимо, однако нагрузки не должны быть чрезмерными, необходима их индивидуальная дозированность, регулярность).
Рекомендуется избегать пищевых продуктов, богатых солями калия (картофеля, изюма и др.), питание больной должно быть рациональным, богатым витаминами, клетчаткой.
Не рекомендуется купаться в холодной воде, есть мороженое и пить холодные напитки.
Важен рациональный выбор профессии (исключение работ, связанных с профессиональной вредностью, переохлаждением, значительными физическими нагрузками, необходимостью быстрой смены позы, работы с заданным ритмом (например, на конвейере).
Рациональное питание
Для нормального усвоения пищи и жизнедеятельности организма больных прогрессирующей мышечной дистрофией необходимо его снабжение всеми пищевыми веществами в определенных соотношениях. Данная категория больных нуждается в специальных диетах, в которых особое внимание должно уделяться консистенции и температуре подаваемых блюд, так как у больных прогрессирующей мышечной дистрофией нарушена функция глотания и затруднено продвижение пищи в желудок, следовательно, пищу необходимо подавать небольшими порциями и средней густоты [Под ред. В.Т. Лапшиной.,2002].
Предпочтение следует отдавать:
— супам-пюре (продукты, используемые для приготовления супов-пюре, после их варки, припускания или тушения протирают сквозь сито, многократно пропускают через мясорубку или измельчают с помощью миксера) из овощей (моркови, шпината, капусты брокколи), птицы (курицы) и рыбы (макрели, лосося, сардины), нерыбных продуктов моря (креветок); чтобы частицы протертых продуктов были равномерно распределены по всей массе и не оседали на дно, рекомендуется добавлять белый соус, приготовленный на отваре овощей);
— блюдам из мяса, рыбы, птицы, приготовленным на пару;
— гарнирам из овощей (моркови, тыквы) в виде пюре;
— блюдам из круп: вязкие каши (рисовая, овсяная);
— блюдам из яиц: яйца всмятку, яичные кашки;
— сладким блюдам: кисели средней густоты (из смородины, облепихи, крыжовника), муссам (из клюквы, ревеня);
— горячим напиткам: зеленый чай [Под ред. Н. А. Шнайдер и др.,2005].
В лечебных учреждениях предпочтение отдают черному чаю, хотя целесообразнее включать в рацион зеленый чай, так как он способствует укреплению кровеносных сосудов, снижает артериальное давление, уровни холестерина и сахара в крови, кроме того, обладает антикариесным, противомикробным и противовирусным действием [Шнайдер Н. А., Шнайдер В. А.,2003].
Зеленый чай. Недавнее исследование на нокаутных мышах показало, что зеленый чай при частом употреблении может иметь терапевтический потенциал для предотвращения гибели (атрофии) миоцитов. Исследователи определили, что потребление, эквивалентное 7 чашкам зеленого чая в день, приводит к уменьшению мышечной слабости в конечностях у лабораторных животных. Окислительный стресс усиливается в мышцах, где экспрессируется аномальный белок дистрофин, поэтому авторы пришли к выводу, что при наследственных заболеваниях, приводящих к синтезу аномального дистрофина или миотонинпротеинкиназы (DMPK) целесообразно употребление зеленого чая как антиоксиданта и ловушки (скавенджера) свободных радикалов, способствующего уменьшению окислительного стресса в скелетной и сердечной мускулатуре. Кроме того, употребление зеленого чая рекомендуется беременным женщинам, страдающим прогрессирующей мышечной дистрофией, как для уменьшения мышечной слабости у самих пациенток, так и для уменьшения степени выраженности дистресс-синдрома и мышечной гипотонии у плода (гипокинезия плода) и новорожденных (синдром вялого ребенка). Несмотря на то что эффективность употребления зеленого чая не исследована у людей, страдающих прогрессирующей мышечной дистрофией, он может быть рекомендован при разработке лечебного питания, поскольку безопасен и не имеет значимых негативных эффектов [Ono S., Takahashi K., Jinnai K. et al.,2003].
Травы. Рекомендуется использовать в виде крутого чая травы, которые настаивают 5–10 мин из листов и цветков и 10–20 мин — из корней, и употреблять 2–4 чашки в день. Можно использовать одну траву или в комплексе несколько трав. При прогрессирующей мышечной дистрофии применяются чаи из следующих трав:
— богатые минералами травы: хвощ полевой (Equisetum arvense L.), крапива двудомная (Urtica dioica), овес посевной (Avena sativa);
— при крампи: цимицифуги корень (Cimicifuga racemosa), калина обыкновенная (Viburnum opulus);
— при миалгии и мышечной слабости: писцидия ярко-красная (Piscidia erythrina), таволга вязолистная (Filipendula ulmaria);
— при кардиальной патологии: боярышник (Crataegus monogyna), розмарин (Rosemarinus officinalis);
— с целью увеличения сопротивления химическим, биологическим и физическим нагрузкам: родиола розовая (Rhodiola rosea) [Griggs R.C., Sansone V., Lifton A., Moxley R.T. III. ,2001].
Маточное молочко. Маточное молочко, или «королевское желе» (Royal Jelly) — естественное вещество, произведенное пчелами для питания пчелиной матки и используемое в качестве пищевой добавки в лечебном питании. Маточное молочко богато витаминами, антиоксидантами, ферментами, гормонами и аминокислотами. Кроме того, оно является естественным антибиотиком и обладает антионкогенным эффектом. Интерес представляет исследование, показавшее целесообразность использования маточного молочка как эффективного средства терапии прогрессирующей мышечной дистрофии и связанных с этим заболеванием поражений мышц скелета и внутренних органов (сердца, желудочно-кишечного тракта, матки и т.п.). Хотя результаты этого исследования нуждаются в уточнении, маточное молочко, являясь веществом натурального происхождения, не имеющим никаких известных серьезных побочных эффектов, может рассматриваться как средство дополнительной терапии из класса витаминов и минералов [Held M., Schneider C., Fleischer K., Jany B.,2001].
Следует помнить, что маточное молочко в нативном состоянии сохраняет биологическую активность лишь несколько дней. Этот срок можно продлить до нескольких месяцев при хранении в герметической упаковке в защищенном от света месте при температуре 0–4 °С. Увеличивает срок хранения лиофилизация, или адсорбция маточного молочка на лактозе. Наиболее эффективным способом является добавление маточного молочка в мед в количестве 0,1–5 % [Aminoff M.J., Beckley D.J., McIlroy M.B. ,2001].
Селен. Селен известен своими антиоксидантными свойствами. Селензависимый фермент глутатион-пероксидаза (GPX) перерабатывает глутатион, уменьшая перекисное окисление липидов посредством каталитической редукции пероксидов, включая свободный пероксид водорода. Селен является кофактором в пределах нескольких метаболических путей, включая GPX-путь, где он присутствует в виде селеноцистеина. Селен метаболизируется в свои биоактивные метаболиты — метилселен и S-метилселеноцистеин, которые, по-видимому, действуют на уровне транскрипционного фактора NFT-[kappa]-B, трансдукции сигналов ключевых пунктов клеточных циклов и запуска апоптоза. Кроме того, селен может занимать важное место как ключевой сигнальный энзим типа тирозинкиназы. Поэтому профилактическая роль селена не ограничена его функцией как антиоксиданта [Шнайдер Н.А., Бахтина Е.А., Макарова Л.Г. и др.,2008].
Дефицит селена играет одну из ключевых ролей при цереброваскулярных заболеваниях и заболеваниях миокарда (в том числе кардиомиопатии) в регионах, где снижено содержание селена в почве и воде. В целом по России, согласно данным эпидемиологических исследований, проведенных в последнее время, более чем у 80 % населения обеспеченность селеном ниже оптимальной [Jiang H., Mankodi A., Swanson M.S. et al. ,2004]. Кроме того, существует взаимообратная связь между уровнем селена в окружающей среде (почве, воде) и возникновением некоторых заболеваний, включая эндемический зоб, синдром внезапной младенческой смерти, рассеянный склероз, некоторые формы кардиомиопатии и шизофрению [Ho T.H., Savkur R.S., Poulos M.G. et al. ,2005].
Форма селена, поступающего с пищей, значительно влияет на всасывание (абсорбцию). L(+)-селенометионин хорошо абсорбируется из желудочно-кишечного тракта и значительно лучше кумулируется в организме по сравнению с неорганическим селеном в форме селенида. L(+)-селенометионин имеет более медленный период полувыведения из организма по сравнению с селенидом. Комплекс селена с метионином обеспечивает более эффективное использование селена [Novelli G., Genarelli M., Menegazzo E. et al. ,2003].
Показано, что концентрация селена в мозге плода снижается к периоду родов и возрастает в постнатальном периоде. Серое вещество головного мозга и гипоталамо-гипофизарная часть мозга имеют наиболее высокое содержание селена [Bassez G., Attarian S., Laforet P. et al. ,2001].
До настоящего времени издано очень мало работ, доступных для анализа, по изучению роли дефицита селена в патогенезе прогрессирующей мышечной дистрофий. При этом заболевании селен может играть роль антиоксиданта, влияющего на уровень свободных радикалов и пероксидов липидов. В некоторых исследованиях показано, что уровень селена в сыворотке крови больных прогрессирующей мышечной дистрофии снижен. В небольших исследованиях показано, что профилактический прием селена с пищей оказывает позитивный эффект на выраженность прогрессирующей мышечной дистрофии. При этом показано уменьшение времени расслабления скелетной мускулатуры на 50 % с незначительным увеличением мышечной силы, но без значимого увеличения объема выполняемой работы [Meola G., Sansone V. ,2000].
Основные традиционные источники селена в питании человека — чеснок, морепродукты, грибы, зерновые, а также мясопродукты. Концентрация селена в них определяется его исходным уровнем в почве и воде, кормах (для продуктов животного происхождения), временем года; она зависит от способа технологической и кулинарной обработки, а также имеет органную и видовую специфичность. Например, морская рыба и другие морепродукты содержат больше этого микроэлемента, чем пресноводная рыба. Содержание селена в мясе и мясопродуктах колеблется от 0,05 до 0,17 мг/кг, в рыбе — от 0,25 до 0,46, в грибах — от 0,55 до 27,9 мг/кг. Селен обнаружен в бразильских орехах, пивных дрожжах, капусте брокколи, буром рисе, красных водорослях, курином мясе, печени, луке, лососе, овощах, зародышах пшеницы и цельных зернах. В той или иной степени накапливать микроэлемент способны около 50 представителей лекарственной флоры, среди которой особое место занимают растения, называемые кумуляторами селена. К пищевым продуктам с особенно низким содержанием селена относят молоко и молочные продукты (от 0,005 до 0,018 мг/кг продукта), крупы, макаронные и кондитерские изделия, овощи и фрукты. В зерновых содержание микроэлемента также не очень велико, но вследствие особенностей характера питания человека их можно считать основными поставщиками селена, особенно если они произрастают на почвах, богатых селеном. Основная форма сeлена в зерне — это Se-Met. По некоторым данным, значительная часть этой аминокислоты сосредоточена в зародыше, поэтому тонкий помол муки с удалением его элементов снижает уровень содержания селена [Шнайдер Н.А., Бахтина Е.А., Макарова Л.Г. и др.,2008].
Перспективными объектами для биотехнологического встраивания селена с целью его дальнейшего использования в пищевых целях являются простейшие грибы, дрожжи и одноклеточные водоросли, в частности спирулина. Обладающая уникальным химическим составом, пищевая микроводоросль спирулина представляет собой очень удобный объект фотобиотехнологии. В Красноярском крае редкостным природным источником спирулины является озеро Плахино (Абанский район). В настоящее время плахинские грязи, богатые спирулиной, используются в лечебных учреждениях г. Красноярска, санаторно-курортном лечении при дерматологической патологии [Шнайдер Н.А., Бахтина Е.А., Макарова Л.Г. и др.,2008].
Одно из важнейших полезных свойств спирулины — антиоксидантное действие, которое может быть существенно усилено путем включения в ее состав биодоступного селена. Другим источником биодоступного селена являются селеносодержащие пищевые дрожжи, крупномасштабное производство которых освоено в настоящее время отечественной промышленностью. Сравнительно низкая себестоимость делает дрожжи очень перспективным и привлекательным пищевым источником органического селена [Day J.W., Ricker K., Jacobsen J.F. et al.,2003].
Витамины. Альфа-токоферол, витамин С, ликопин и бета-каротин значительно улучшают диетическую усвояемость селена, так как имитируют приближающийся к идеалу пищевой комплекс. Жирорастворимые витамины значительно повышают степень абсорбции селена. Так, бета-каротин увеличивает всасываемость селена в тонком кишечнике в 1,8 раза. В настоящее время доказано наличие синергизма в плане биодоступности селена для витамина А, аскорбиновой кислоты, бета-каротина, фосфолипидов и ликопина. Наличие всех компонентов, действующих синергически, позволяет получить максимально возможную биодоступность как для селена, так и для остальных компонентов этого антиоксидантного комплекса. Введение для коррекции селена препарата селенопирана в комплексе с альфа-токоферолом и аскорбиновой кислотой при нагрузке метионином повышает содержание восстановленного глутатиона на 20–70 %, активность антиоксидантных ферментов на 20–50 % и снижает содержание продуктов перекисного окисления липидов на 10–60 % в раннем постнатальном и старческом периоде. Фосфолипиды наряду с жирорастворимыми витаминами комплекса повышают биоусвояемость селена и его транспортную доставку к тканям и органам, богатым липидами (мозг, проводящие нервные пути, спинномозговая жидкость, печень, мембраны клеток) [Bassez G., Attarian S., Laforet P. et al. ,2001; Спиричев В.Б.,2003].
Сбалансированная диета с высоким содержанием свежих овощей, фруктов, растительных масел и продуктов из цельного зерна, действительно, может полностью обеспечить физиологические потребности организма больных прогрессирующей мышечной дистрофией в основных природных антиоксидантах, аскорбиновой кислоте, токоферолах, каротиноидах и многих других минорных компонентах, вносящих свой вклад в систему антиоксидантной защиты. Однако реальный пищевой рацион преобладающего большинства пациентов, не имеющих национальной привычки и возможности употреблять большое количество перечисленных выше продуктов, крайне беден витамином С, каротиноидами и далеко не всегда оптимально обеспечен витамином Е [Fardaei M., Rogers M.T., Thorpe H.M. et al. ,2002].
Поскольку быстро изменить сложившуюся структуру и привычки питания больных прогрессирующей мышечной дистрофией и ликвидировать отрицательные последствия реально существующего, широко распространенного и глубокого дефицита ряда биоантиоксидантов (аскорбиновой кислоты, 3-каротина, селена, а в ряде случаев и витамина Е) невозможно, возникает необходимость кардинальной перестройки рациона в сторону существенного увеличения потребления богатых антиоксидантами продуктов питания [Fardaei M., Rogers M.T., Thorpe H.M. et al. ,2002].
Аминокислоты
Таурин. Таурин необходим для нормального обмена натрия, калия, кальция и магния. Проведены исследования на пациентах с прогрессирующими мышечными дистрофиями и на здоровых людях до и после парентерального введения таурина. Изменения возбудимости m.thenari было связано с уровнем калия и хлоридов в сыворотке крови. Фактическая концентрация электролитов была сопоставлена с ожидаемой, при условии, что вводимые внутривенно электролиты не транспортировались в клетки-мишени. Ожидаемые концентрации были вычислены эмпирически. Мышцы у пациентов с прогрессирующей мышечной дистрофией оказались очень чувствительны к внеклеточному калию в отличие от мышц здоровых людей и были неспособны аккумулировать вызванную калием повышенную возбудимость мышц. Таким образом, таурин способен увеличивать внутриклеточную концентрацию калия и мембранную проводимость. Таурин содержится в ряде пищевых продуктов — яйцах, рыбе, мясе, моллюсках, птице, но не встречается в белках растительного происхождения [Лобзин В.С. и др.,2002].
Кофермент Q10. Кофермент Q10 (CoQ10), или убихинон, — мощный антиокислитель и митохондриальный дыхательный кофактор. Он обладает мембраностабилизирующим эффектом. Митохондрии производят энергию, которая расходуется в процессе жизнедеятельности. Убихинон действует так, чтобы облегчить сложный ряд реакций, которые происходят в пределах митохондрий. Химические реакции, в конечном счете, поставляют энергию, которая может быть сохранена для более позднего использования или израсходована в данный момент. В синтезе кофермента Q10 участвует селен [Folkers K; Simonsen R, 1995; Hund E., Jansen O., Koch M. et al. ,2001].
Учитывая важность всех этих процессов, ученые задались вопросом, может ли дополнительное введение убихинона улучшить состояние больных прогрессирующей мышечной дистрофией, которые страдают от снижения мышечной силы и несовершенного метаболизма энергии в пределах миоцитов. Пациенты с самой большой степенью генетической мутации имеют самые низкие уровни кофермента Q10. Это говорит о том, что дефицит кофермента Q10 действительно связан с несовершенным энергетическим обменом миоцитов при прогрессирующей мышечной дистрофии [Moxley R.T. IIIrd, Udd B., Ricker K. ,2000].
Наибольшее количество кофермента Q10 обнаружено в продуктах питания животного происхождения, таких как мясо, печень, бычье сердце и т.д. Также кофермент содержится в макрели, лососе, сардине, арахисе, шпинате. Поскольку кофермент Q10 является жирорастворимым веществом, его рекомендуют использовать вместе с продуктами, содержащими жир, такими как сыр, масло и др. Лучше всего употреблять с небольшим количеством витамина Е, так как это помогает предохранению кофермента Q10 от разрушения [Лобзин В. С., Сайкова Л. А., Шиман А. Г.,2000].
Список используемой литературы
1. Бадалян Л. О., Темин П.А., Калинин В.А. и др. Прогрессирующая миодистрофия с контрактурами и злокачественным течением – вариант болезни Эмери-Дрейфуса Журн. невропатологии и психиатрии 90:3, 1990
2. Бадалян Л.О. Невропатология, М., 2008
3. Бадалян Л.О., Скворцов И.А. Клиническая электронейромиография, М., 1986
4. Баранов В.С. Соросовский образовательный журнал, № 3, 1999
5. Белозеров Ю.М., Никанорова М.Ю., Перминов В.С., Страхова О.С. Прогрессирующая мышечная дистрофия Эмери-Дрейфуса. Журн. Альманах клинической медицины. Актуальные вопросы практической неврологии, № 4, М., 2001.
6. Вельтищев Ю.Е., Темин П.А. Наследственные болезни нервной системы. — М.: Медицина, 1998.
7. Гаусманова - Петрусевич И. Мышечные заболевания. Польск. Госуд. Мед. Изд. - Варшава, 2001.
8. Гехт Б.М. и Ильина Н.А. Нервно-мышечные болезни, М., 1998.
9. Гехт Б.М., Касаткина Л.Ф., Самойлов М.И., Санадзе А.Г. Электромиография в диагностике нервно-мышечных заболеваний. - Таганрог: Изд. ТРТУ., 1997.
10. Горбунова В.Н., Баранов B.C. Введение в молекулярную диагностику и генотерапию наследственных заболеваний. Ст-Петербург 1997.
11. Горбунова В.Н., Савельева Е.А., Красильников В.В. Молекулярная неврология. — СПб.: Интермедика, 2000. — Ч. 1.
12. Гринио Л.П. Атлас нервно-мышечных болезней - М.: Издат. дом АНС, 2004.
13. Гринио Л.П. Дюшенновская миодистрофия. - Н.Новгород: Изд-во НГМА., 1998
14. Гринио Л.П., Агафонов Б.В. Миопатии. — М.: Медицина, 1997.
15. Гусев Е.И., Коновалов А.Н., Скворцова В.И., Гехт А.Б. Неврология, 2009.
16. Гусев Е.И., Никифоров А.С. Общая неврология, 2007
17. Евграфов О.В., Макаров В.Б. ДНК-диагностика наследственных заболеваний. Итоги науки и техники: Генетика человека 9; 1991.
18. Евтушенко С.К., Евтушенко И.С., Новые современные технологии в терапии нейромышечных заболеваний, направленные на замедление их прогрессирования. Международный неврологический журнал № 4(26) 2008.
19. Евтушенко С.К., Садеков И.А. Наследственные заболевания и пороки развития нервной системы в практике невропатолога. — Донецк: Лебедь, 1994.
20. Зенков Л. Р., Ронкин M. А. Функциональная диагностика нервных болезней. - M., 1991.
21. Иллариошкин С. Н., Иванова-Смоленская И.А., Маркова Е.Д. ДНК- диагностика и медико-генетическое консультирование в неврологии. М., 2002
22. Иллариошкин С.Н., Иванова-Смоленская И.А. Молекулярные основы прогрессирующих мышечных дистрофий Журнал неврологии и психиатрии. — 1998. — № 10.
23. Казаков В.М. Клинико-молекулярно-генетическая классификация мышечных дистрофий (научный обзор с комментариями) Неврол. журнал. — 2001 — № 3.
24. Карпищенко А.И. Медицинские лабораторные технологии. Ст-Петербург: Интермедика 1999.
25. Карпович Е.И., Казакова Л.В., Колбасова Л.В. и др. Случай прогрессирующей мышечной дистрофии Эмери—Дрейфуса. Журн неврол психиат 98: 10, 1998.
26. Крахмалева И.Н., Липатова Н.А., Шишкин С.С. и др. Генетика 1999.
27. Лечение нервных болезней, под ред. В.К. Видерхольда, пер. с англ., М., 2004
28. Лобзин В. С., Сайкова Л. А., Шиман А. Г. Нервно-мышечные болезни. — СПб.: Гиппократ, 2000. — С. 138—144.
29. Лобзин В.С. и др. Восстановительная и корригирующая терапия нервно-мышечных заболеваний, Л., 2002.
30. Максимова Н. Р. И. А. Николаева М. Н. Коротов Т. Икеучи О. Онодера М. Нишизава С. К. Степанова Х. А. Куртанов А. Л. Сухомясова А. Н. Ноговицына Е. Е. Гуринова В. А. Степанов В. П. Пузырев Клинико-генеалогическая и молекулярно-генетическая характеристика окулофарингеальной дистрофии в республике Саха (Яуктия) журн. Неврологии и психиатрии им. С.С. Корсакова №6, 2008
31. Мальмберг С.А. и соавт. Конечностно-поясная мышечная дистрофия: случай ранней детской формы у 2 сибсов. Практическая неврология. - М., 2001.
32. Мальмберг С.А., Петрухин А. С., Широкова В.И. Мышечная дистрофия Эмери—Дрейфуса. Неврол журн, 1, 2000
33. Новиков П.В., О.В. Евграфов ДНК-диагностика наследственных заболеваний у детей в Российской Федерации: состояние и проблемы Российский вестник перинатологии и педиатрии, N5,1999
34. Под ред. В.Т. Лапшиной. Сборник рецептур блюд и кулинарных изделий диетического питания / М.: Хлебпродинформ, С. 632.2002.
35. Под ред. Н. А. Шнайдер и др. Миотония: Руководство для врачей /— М.: НМФ "МБН", 2005.
36. Руденская Г.Е., Тверская С.М., Чухрова А.Л. и др. Разнообразие болезней, обусловленных мутациями гена LMNA. Мед генетика 2004.
37. Самуэльс М. Неврология, 1997
38. Свердлов Е.Д. Молекулярная генетика, микробиология и вирусология, 1997
39. Спиричев В.Б. Витамины-антиоксиданты в профилактике и лечении сердечно-сосудистых заболеваний. Витамин Е // Вопр.питания. 2003. № 6. С. 45—51.
40. Страхова О.С., Белозерова Ю.М., Темин П.А. Кардиомиопатия при прогрессирующей мышечной дистрофии Дюшенна, 1999.
41. Сухомясова А.Л. Аутосомно-доминантная миотоническая дистрофия в Республике Саха (Якутия): Автореф. дис. канд. мед. наук. Томск, 2005.
42. Тверская С.М., Руденская Г.Е., Чухрова А.Л., Поляков А.В. ДНК-диагностика прогрессирующей мышечной дистрофии Эмери-Дрейфуса. Журн неврол психиат; 103: 6, 2003.
43. Темин П.А., Белозеров Ю.М., Никанорова М.Ю., Страхова О. С. Прогрессирующая мышечная дистрофия Эмери—Дрейфуса. В кн.: Наследственные болезни нервной системы. Под ред. Ю.Е. Вельтищева, П.А. Темина. М: Медицина 1998.
44. Тетенев Ф.Ф., Бодрова Т.Н., Емельянова Н.В. Биомеханика дыхания у больных с прогрессирующими мышечными дистрофиями Журн. неврол. и психиатрии.— № 8— 2000.
45. Умаханова Р. С. С. Жилина Г. Р. Мутовин Клинический полиморфизм прогрессирующей мышечной дистрофии Эрба – Рота. журн. Неврологии и психиатрии им. С.С. Корсакова №9, 2005
46. Шаховская Н.И. Генетическая гетерогенность миопатии Дюшенна - Беккера и организация медицинской помощи детям с этим заболеванием в Московском регионе: Автореф. дис. канд.мед. наук. М., 2000.
47. Шишкин С.С. Наследственные нервно-мышечные болезни. М: Изд-во ВИНИТИ, 1997.
48. Шишкин С.С. Современные представления о механизмах реализации генетической информации и их нарушениях при некоторых видах менделирующих болезней. В сб.: Теоретические и прикладные проблемы медицинской генетики. М: Изд-во МОИП 1998.
49. Шишкин С.С., Калинин В.Н. Медицинские аспекты биохимической и молекулярной генетики. М: Изд-во ВИНИТИ, 1999.
50. Шишкин С.С., Ковалев Л.И. Журн невропатол и психиат №3;1998.
51. Шишкин С.С., Н.И. Шаховская, И.Н. Крахмалева Клинический полиморфизм, генетическая гетерогенность и проблемы патогенеза первичных миопатий Неврология, №2,2002.
52. Шишкин С.С., Шаховская Н.И., Лунга И.Н. и др. Наследственные нервно-мышечные заболевания, некоторые проблемы оказания помощи больным и отягощенным семьям. В кн.: Многоликость современной генетики человека. Под ред. С.С. Шишкина. М - Уфа: Изд-во Гилем 2000.
53. Шнайдер Н.А., Бахтина Е.А., Макарова Л.Г. и др. Роль селена в питании больных с дистрофической миотонией // Вестник НГУ. — 2008. — № 1. — С. 91-96.
54. Шток В.Н. Фармакотерапия в неврологии, 2003
55. Яхно Н.Н., Штульмен Д.Р., Мельничук П.В. Болезни нервной системы: В 2 т. — М.: Медицина, 2001.
56. Adzija D. et al - A prospective cardiological study in patients with progressive muscular dystrophy Duchenne type in Belgrade - Acta-Cardiomiologica- 1994.
57. Ahn A.H., Kunkel L.M. The structural and functional diversity of dystrophin Nature Genet. — 1998.
58. Aminoff M.J., Beckley D.J., McIlroy M.B. Autonomic function in myotonic dystrophy // Arch. Neurol. — 2001. — Vol. 42. — P. 16.
59. Backman E., Henriksson K.G. Neuromusc Disorders, 1995.
60. Bashir R. et al. A gene for autosomal recessive limbgirdle muscular dystrophy maps to chromosome 2. Hum. Molec.Genet. 1994.
61. Bashir R., Britton S., Strachan T. et al. Nat Genet 20; 1998.
62. Bassez G., Attarian S., Laforet P. et al. Proximal myotonic myopathy (PROMM): clinical and histology study // Rev. Neurol. — 2001. — Vol. 157. — P. 209-218.
63. Beauchamp J.R. Morgan J.E., Pagel C.N. et al J. Cell Biol, 1999.
64. Becher M. W., Morrison L., Davis L.E. et al. Oculopharyngeal Muscular Dystrophy in Hispanic New Mexicans. JAMA 2001.
65. Bejaoui K., Hirabayashi K., Hentati F. et al. Neurology 1995.
66. Blumen S.C., Brais B., Korczyn A.D. et al. Homozygotes for oculopharyngeal muscular dystrophy have a severe form of the disease. Ann Neurol 1999.
67. Bushby K.M.D. et al - The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy - J.Neurol.-1993.
68. Chenard A.A. et al - Ventricular arrhythmia in Duchenne muscular dystrophy: Prevalence, significance and prognosis – Neuromuscular-disord. – 1993.
69. Codere F., Brais B., Rouleau G., Lafontaine E. Oculopharyngeal muscualr dystrophy: What's new? Orbit 2001.
70. Comi G.P. et al – Clinical variability in Becker muscular dystrophy. Genetic, biochemical and immunohistochemical correlates - Brain 1992.
71. Day J.W., Ricker K., Jacobsen J.F. et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum // Neurol. — 2003.— Vol. 60, № 4.— P. 657-664.
72. Emery A.E.H. Diagnostic Criteria for Neuromuscular Disorders. European Neuromuscular Centre. The Netherlands 1994.
73. Fardaei M., Rogers M.T., Thorpe H.M. et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells // Hum. Mol. Genet. — 2002. — Vol. 11. — P. 805-814.
74. Folkers K; Simonsen R Two successful double-blind trials with coenzyme Q- 10 (vitamin Q-10) on muscular dystrophies and neurogenic atrophies. Biochimica-et- Biophysica-Acta-Molecular-Basis-of-Disease, 1995.
75. Griggs R.C., Sansone V., Lifton A., Moxley R.T. III. Hypothyroidism unmasking proximal myotonic myopathy (PROMM) // Neurology. — 2001. — Vol. 48. — P. 229.
76. Gussoni, E, Soneoka, Y., Strickland, C. D et al. Nature, 1999.
77. Held M., Schneider C., Fleischer K., Jany B. A patient with muscle pain after a journey to the tropics. Myocardial involvement in proximal myotonic myopathy // Dtsch. Med. Wochenschr. — 2001. — Vol. 123. — P. 1201-1206.
78. Ho T.H., Charlet B.N., Poulos M.G. et al.Muscleblind proteins regulate alternative splicing // EMBO J. — 2004. — Vol.23. — P.3103-3112.
79. Hoffmann E.P., Kunkel L.M., Angelini C. et al. Neurology 2009.
80. http:// doctor.ru/medinfo
81. Hund E., Jansen O., Koch M. et al. Proximal myotonic myopathy with MRI white matter abnormalities of the brain // Neurology. — 2001. — Vol. 48. — P. 33-37.
82. Ishikawa Y, Bach JR, Sarma RJ et al., Cardiovascular consideration in the management of neuromuscular disease. Seminars in neurology,1995.
83. Jiang H., Mankodi A., Swanson M.S. et al. Myotonic dystrophy type 1 associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins, and deregulated alternative splicing in neurons // Hum. Mol. Genet. — 2004. — Vol. 13. — P.3079-3088.
84. Kaplan J.C., Fontaine В. Neuromuscular disorders: gene location. Neuromusc Disord 1999.
85. Kay M., Liu D, Hoogerbrugge P.M. Proc. Natl. Acad. Sci. USA, 94, 1997.
86. Khurana T.S., Prendergast R.A., Alameddine H. et al. J Exp Med 1995.
87. Le-Thiet-Thanh; Nguyen-Thi-Man; Hori-S; Sewry-CA; Dubowitz-V; Morris-GE Characterization of genetic deletions in Becker muscular dystrophy using monoclonal antibodies against a deletion-prone region of dystrophin. Am-J-Med-Genet. 58(2), 1995.
88. Lim L.E., Duclos P., Broux O. et al. Nat Genet 11;1995.
89. Liu J., Aoki M., Illa I. et al. Nat Genet 20;1998.
90. Maeda M; Nakao S; Miyazato H; et al. Cardiac involvement in Becker muscular dystrophy J-am-coll-cardiol. 22/7, 1996.
91. McKusik V., Amberger J. The morbid anatomy of the human genome chromosomal location causing disease J. Med. Genet. 2003.
92. McNally E., Passos-Bueno R., Bonnemann C.G. et al. Am J Hum Genet 1996.
93. Meola G. Clinical and genetic heterogeneity in myotonic dystrophies // Muscle Nerve — 2000. — Vol. 23. — P. 1789-1799.
94. Minetti C., Sotgia F., Bruno C. et al. Nat Genet 18; 1998.
95. Mirabella M., Silvester G., Rosa G. et al. GCG genetic expansions in Italian patients with oculopharyngeal muscular dystrophy. Neurology 2000.
96. Moreira E., Vainzof M., Marie S. et al. Am J Hum Genet 61; 1997.
97. Moxley R.T. IIIrd, Udd B., Ricker K. Proximal myotonic myopathy (PROMM) and other proximal myotonic syndromes // Neuromuscul. Disord. — 2000. — Vol. 8. — P. 519-520.
98. Muntoni F., Mateddu A., Marchei F. et al. J Neurol Sci l20; 1993.
99. Nevo Y., Muntoni F., Sewery C. et al. Neuromusc Disorders 1998.
100. Nigro V., de Sa Moreira E., Piluso G. et al. Nature Genet 14; 1996.
101. Novakovic I., Todorovic S., Apostolski S. et al. Neuromusc Disord 1998.
102. Novelli G., Genarelli M., Menegazzo E. et al. (CTG)n triplet mutation and phenotype manifestations in myotonic dystrophy // Biochem. Med. Metab. Biol. — 2003. — Vol. 50. — P. 85-92.
103. Ono S., Takahashi K., Jinnai K. et al. Loss of serotonin-containing neurons in the raphe of patientswith myotonic dystrophy: a quantitative immunohistochemical study and relation to hypersomnia // Neurology. — 2003. — Vol. 50. — P. 535-538.
104. Palmucci L., Doriguzzi C., Mongini T. et al. Neurology 2004.
105. Partridge T., Lu Q.L., Morris G., Hoffman E Nature Medicine, 4, 1998
106. Penninger J.M., Neu N., Bachmaier К. The Immunologist 1996.
107. Piantadosi C., Nigro V., Servider S. et al. Neuromusc Disorders 1998.
108. Piccolo F., Roberds S.L., Jeanpierre M. et al. Nat Genet 10; 1995.
109. Porter J.D. Neuromusc Disord 1998.
110. Quinlivan-RM; Dubowitz-V Cardiac transplantation in Becker muscular dystrophy. Neuromuscul-Disord. 1992.
111. Rando T.A., Dziesietnik M.H., Dhawan J. et al. Neurology 1997.
112. Rees W; Schuler S; Hummel M; Hetzer R - Heart transplantation in patients with muscular dystrophy associated with end-stage cardiomyopathy J-heart- lung-transplant. 12/5, 1993.
113. Richard I., Brenguier L., Dincer P. et al. Am J Hum Genet 60; 1997.
114. Saito K.et al - Molecular genetic analysis of Duchenne/Becker muscular dystrophy families. In: Сlin-neurol. - 2002.
115. Sharma K.R., Mynhies M.A., Robert Y., Miller R.Y. Neurology 1993.
Похожие рефераты:
Литература - Другое (книга по генетике)
Аллергия и аллергические заболевания
Литература - Другое (клиника, диагностика, лечение некоторых форм)
Массаж при патологии дыхательной системы
Заболевания периферической нервной системы
Общая нозология. Типовые патологические процессы
Акушерство. Методические рекомендации кафедры
Исследование соотношения в мышцах С- и Х-белков в норме и при патологии
Диагностика и консервативное лечение асимметрии таза у детей