| Скачать .docx | Скачать .pdf |
Реферат: Прионные болезни человека
О Г Л А В Л Е Н И Е
Историческая справка…………………………………………………………………... 2
Понятия и термины……………………………………………………………………... 5
Современная классификация прионных болезней……………………………………. 6
Место прионных болезней в инфекционной патологии……………………………… 7
Структура прионных белков…………………………………………………………… 9
Физико-химические свойства прионов………………………………………………... 11
Биологические особенности прионов………………………………………………….. 13
Прионный ген…………………………………………………………………………… 14
Патология центральной нервной системы при прионных болезнях человека ……... 17
Изучение прионных болезней………………………………………………………….. 26
Лечение и профилактика прионных болезней человека……………………………… 29
Диагностика прионных болезней человека и индикация прионного белка………… 31
Заключение………………………………………………………………………………. 33
Список литературы……………………………………………………………………… 34
ИСТОРИЧЕСКАЯ СПРАВКА
Проблема прионных болезней родилась в рамках учения о медленных инфекциях, когда в 1954 г. B.Sigurdsson изложил результаты своих многолетних исследовании массовых заболеваний среди овец, завезенных на о. Исландия из Германии в 1933 г. для развития каракулеводства. Несмотря на явные клинические различия и неодинаковую локализацию повреждений органов и тканей, B.Sigurdsson сумел обнаружить среди изученных им заболеваний принципиальное сходство, которое в современном виде может быть суммировано в виде четырех главных признаков, отличающих медленные инфекции:
• необычно продолжительный (месяцы и годы) инкубационный период;
• медленно прогрессирующий характер течения;
• необычность поражения органов и тканей;
• неизбежность смертельного исхода.
Среди изученных B.Sigurdsson заболеваний овец была подробно исследована давно и хорошо знакомая во многих странах болезнь этих животных, известная под названием "скрепи".
Три года спустя в противоположном регионе – на о. Новая Гвинея – D.C.Gajdusek и V.Zigas (1957) обнаружили и описали новое заболевание среди папуасов-каннибалов, которое известно сегодня под названием "куру". Болезнь носила массовый характер и вскоре была доказана ее инфекционная природа.
Благодаря исследованиям W.Hadlow (1959) было выявлено большое сходство между клиническими проявлениями, эпидемиологическими показателями и патоморфологической картиной куру у человека и скрепи у овец, на основании чего стало очевидным, что медленные инфекции могут поражать не только животных, но и людей.
Массовый характер медленных инфекций, естественно, ставил вопрос об их этиологии, и начавшиеся энергичные поиски в этом направлении вскоре принесли свои плоды.
За короткое время был накоплен большой и весьма неожиданный фактический материал: оказалось, что очень многие вирусы, давно и хорошо известные как возбудители острых заболеваний, способны при определенных условиях вызывать в организме медленный инфекционный процесс, полностью отвечающий всем четырем признакам медленных инфекций. В числе таких вирусов вскоре оказались вирусы кори, краснухи, герпеса, клещевого энцефалита, лимфоцитарного хориоменингита, африканской лихорадки свиней, инфекционной анемии лошадей, бешества, вирусы семейства папова, гриппа, иммунодефицита человека и др.
Следует особо подчеркнуть, что, начиная с сообщений B.Sigurdsson, в литературе постепенно накапливались данные об особой группе медленных инфекций человека и животных, патоморфологические признаки которых весьма существенно отличались выраженным своеобразием, проявляющимся только в поражении центральной нервной системы, в которой на основе первично-дегенеративных процессов (без признаков воспаления) развивается характерная картина формирования так называемого губкообразного состояния серого и/или белого вещества головного, а иногда и спинного мозга, что может сопровождаться образованием амилоидных бляшек и выраженным глиозом.
Подобное своеобразие патоморфологической картины определило и первичное (и до сих пор используемое) название всей группы этих необычных болезней – "трансмиссивные губкообразные энцефалопатии" (ТГЭ). Именно трансмиссивность губкообразных изменений только в центральной нервной системе и является их патогномоничным признаком.
На протяжении нескольких десятилетий все попытки обнаружить возбудителей ТГЭ заканчивались неудачей, хотя инфекционная природа их была точно доказана многочисленными опытами передачи заболеваний различным животным и не вызывала сомнений.
Вместе с тем на протяжении этих лет накапливались данные, которые не прямо, но косвенно позволяли судить по крайней мере о некоторых свойствах возбудителей ТГЭ. И первые же положительные результаты подтвердили правильность или во всяком случае эффективность вирусологического подхода в изучении этиологии ТГЭ.
Не имея возможности работать с самим этиологическим агентом, исследователи предприняли разностороннее изучение инфицированной мозговой ткани, наивысшее содержание инфекционного агента в которой было давно установлено. При этом оказалось, что предполагаемый инфекционный агент обладает следующими свойствами:
• способен проходить через бактериальные фильтры с диаметром пор от 25 до 100 нм;
• не способен размножаться на искусственных питательных средах;
• воспроизводит феномен титрования (вызывает гибель зараженных животных при высоких значениях ИД5о);
• накапливается до титров 105 - 1011 на 1 г мозговой ткани;
• способен первоначально репродуцироваться в селезенке и других органах ретикулоэндотелиальной системы, а затем в мозговой ткани;
• способен к адаптации к новому хозяину, что нередко сопровождается укорочением инкубационного периода;
• характеризуется наличием генетического контроля чувствительности у некоторых хозяев (например, у овец и мышей для возбудителя скрепи);
• имеется специфический круг хозяев для данного штамма возбудителя;
• может регистрироваться изменение патогенности и вирулентности у разных штаммов для различного круга хозяев;
• возможна селекция штаммов из дикого типа;
• возможно воспроизведение феномена интерференции (например, медленно репродуцирующегося штамма возбудителя скрепи с быстро репродуцирующимся штаммом в организме мышей);
• возможна персистенция в культуре клеток, полученных из органов и тканей зараженного организма.
Однако наряду с приведенными признаками у возбудителей ТГЭ были обнаружены свойства, которые отличались от таковых у известных вирусов. Так, возбудители ТГЭ оказались устойчивыми к действию бета-пропиолактона, формальдегида, глутаральдегида, ЭДТА, нуклеаз (РНКазы А и III, ДНКазы I), псораленов, нагревания до 80°С (при неполной инактивации в условиях кипячения), УФ-лучей, ионизирующей радиации, ультразвука. Более того, ни одним из инфекционных материалов, полученных от животных или людей, погибших от ТГЭ, долгое время не удавалось заразить интактные клеточные культуры.
Перечисленные своеобразные свойства дали основание рассматривать возбудителей ТГЭ как "необычные вирусы". В связи с этим и сами заболевания некоторое время подразделяли на две группы: медленные инфекции, вызываемые обычными вирусами, и медленные инфекции, вызываемые необычными вирусами.
Однако в начале 80-х годов эти нечеткие определения были значительно конкретизированы, а в дальнейшем и уточнены, что целиком и полностью связано с успехами в расшифровке природы возбудителей ТГЭ. Американский биохимик Стэнли Прузинер, используя новые подходы к накоплению и очистке инфекционного начала в мозговой ткани зараженных хомяков, показал, что возбудителем наиболее распространенной в природе ТГЭ – скрепи (заболевание, которое в природе встречается среди овец и коз) – является безнуклеиновый низкомолекулярный (27–30 кДа) белок, который он назвал "инфекционный прионный белок". В качестве инфекционной единицы С. Прузинер предложил наименование "прион". Термин "прион" образован как анаграмма английских слов "белковая инфекционная (частица)" – "proteinaceousinfectious (particles)".
Прион как инфекционная единица состоит из молекул инфекционного прионного белка.
Результаты исследований последних 15 лет полностью подтвердили прионную природу возбудителей ТГЭ, и на этом основании эти заболевания обозначают теперь как "прионные болезни.
ПОНЯТИЯ И ТЕРМИНЫ
Понятия и термины. Постепенное накопление фактов, все более полно характеризующих особенности прионных болезней и их возбудителей, естественно, порождали появление новых терминов и понятий, которые, конечно же, будут использованы при дальнейшем изложении материалов в этой книге и поэтому нуждаются в специальном объяснении.
Прион – малая белковая инфекционная частица, устойчивая к инактивирующим воздействиям, которые модифицируют нуклеиновые кислоты. Прионы по большей части или исключительно состоят из молекул инфекционного прионного белка и вызывают ТГЭ у человека и животных.
PrP – прионный белок.
PrPSc _ инфекционный прионный белок, который вызывает скрепи (scrapie) у овец и коз и другие прионные болезни животных и человека. Однако, учитывая, что скрепи является наиболее распространенной в природе прионной болезнью, для обозначения инфекционности прионного белка использованы первые буквы названия заболевания – "Sc" (scrapie).
PrPC – неинфекционный прионный белок, который носит наименование "клеточный" и в этом случае "С" – начальная буква английского слова cell (клетка). Неинфекционный (клеточный) прионный белок является жизненно необходимым белком, обнаруживаемым в организме всех млекопитающих, включая и человека. Одной из отличительных черт клеточного прионного белка является его высокая чувствительность к переваривающему действию протеазы К, под действием которой PrPC полностью разрушается.
PrP 27 -30 – инфекционный прионный белок, сохраняющийся в результате переваривающего воздействия протеазы К на исходный инфекционный прионный белок PrPSc . Его молекулярная масса в результате гидролитического воздействия протеазы К снижается лишь незначительно и сохраняется на уровне 27–30 кДа.
PRNP– ген, кодирующий синтез клеточного прионного белка (PrPC в организме человека, локализованный на хромосоме 20.
Prnp – ген, кодирующий синтез клеточного прионного белка (PrPC в организме мыши, локализованный на хромосоме 2.
Прионные палочки – белковые структуры, выявляемые в мозговой ткани зараженных животных или человека и представляющие собой главным образом или исключительно агрегированные молекулы инфекционного прионного белка (PrP 27–30), сформированные в результате экстракции детергентами и ограниченного протеолиза исходного инфекционного прионного белка (PrPSc ). Морфологически и гистохимически прионные палочки неотличимы от многих амилоидных структур.
PrP-амилоидные бляшки – амилоидные бляшки, состоящие из прионного белка, обнаруживаемые в мозговой ткани животных или людей, погибших от прионных болезней.
Конформационные белки – белки, у которых в результате изменений третичной или даже четвертичной структуры меняются некоторые свойства.
СОВРЕМЕННАЯ КЛАССИФИКАЦИЯ
ПРИОННЫХ БОЛЕЗНЕЙ
Как и любая другая, классификация прионных болезней представляет собой попытку искусственного группирования объектов с целью систематизации фактического материала для простоты его восприятия, обоснованности обобщений и эффективности дальнейших исследований хотя бы в ближайшей перспективе. Отсюда понятно, что большие успехи, достигнутые за последние 10 – 15 лет в области изучения прионов и вызываемых ими заболеваний, обосновали естественную потребность в систематизации накопленных данных.
Таблица 1. Современная классификация прионных болезней человека и животных |
|
Нозологическая форма |
Естественный хозяин |
| Болезнь Крейтцфельдта – Якоба | Человек |
| Куру | - // - |
| Синдром Герстманна – Штреусслера – Шейнкера | - // - |
| Фатальная семейная инсомния (смертельная семейная бессонница) | - // - |
| Скрепи | Овцы и козы |
| Трансмиссивная энцефалопатия норок | Норки |
| Хроническая изнуряющая болезнь | Олени и лоси |
| Губкообразная энцефалопатия крупного рогатого скота | Коровы и быки |
| Губкообразная (спонгиоформная) энцефалопатия кошек | Кошки |
Губкообразная энцефалопатия экзотических копытных |
Антилопы и большой куду |
Список прионных болезней человека возглавляет болезнь Крейтцфельдта–Якоба, которая хронологически хотя и была включена в число инфекционных ТГЭ позднее куру, тем не менее является как бы основным заболеванием, в то время как куру и синдром Герстманна–Штреусслера–Шейнкера рассматриваются как особые ее формы.
Среди прионных болезней животных основным заболеванием является скрепи в связи с тем, что именно эта болезнь рассматривается как прототип всех прионных болезней человека и животных. Указанное выше удвоение числа прионных болезней животных связано с разразившейся с 1986 г. в Великобритании эпизоотией губкообразной энцефалопатии крупного рогатого скота (ГЭКРС).
Детальные исследования условий передачи прионных болезней у людей позволили в самое последнее время предложить еще один вариант классификации именно этой немногочисленной группы заболеваний, основанный на характере и особенностях их возникновения. Установлено, что в отличие от всех известных инфекционных заболеваний прионные болезни человека могут возникать как:
1) инфекционные,
2) спорадические,
3) наследственные.
МЕСТО ПРИОННЫХ БОЛЕЗНЕЙ В ИНФЕКЦИОННОЙ ПАТОЛОГИИ
Место прионных болезней в инфекционной патологии человека и животных определяется особенностями, присущими этим заболеваниям.
Первая из них связана с необычностью возбудителей, свойства которых резко отличают их от всех известных инфекционных агентов. Именно это обстоятельство выделяет прионные болезни в особую категорию болезней, абсолютно "безразличных" к средствам как лекарственной терапии, так и к разнообразным средствам и методам иммунотерапии. Эти особенности заставляют переносить основное внимание в борьбе с прионными болезнями на меры предупредительные, нежели лечебные. Хотя справедливости ради заметим, что даже абсолютная фатальность прионных болезней не может и не должна служить основанием для прекращения поисков эффективных лекарственных средств. Именно поэтому в 1998 г. в Москве на V Российском конгрессе "Человек и лекарство" был организован и с успехом проведен специальный симпозиум, целиком посвященный прионным болезням человека и животных.
Что же касается средств иммунотерапии и, естественно, иммунопрофилактики, то здесь пока не существует реальных оснований, которые позволяли бы рассчитывать на успех по крайней мере в обозримом будущем, в связи с тем что инфекционный прионный белок PrPSc иммунологически не отличим от нормального прионного белка PrPC .
Вторая особенность прионных болезней обусловлена тем, что они представляют собой неотъемлемую часть теперь уже достаточно обширной (около 40 нозологических форм) группы медленных инфекций человека и животных. Как известно, подавляющее большинство этих заболеваний вызывают вирусы, известные как возбудители острых инфекций. Это лишний раз подчеркивает справедливость утверждения о том, что большинство вирусов в зависимости от условий заражения (или пребывания) способствует развитию в организме различных форм инфекционного процесса.
В связи с этим прионные болезни занимают особое положение, так как их возбудители не способны к столь выраженной универсальности, как у вирусов, и они (инфекционные прионные белки -PrPSc ) не формируют и не поддерживают в организме иные процессы, кроме медленного и (как это было установлено уже давно и впоследствии неоднократно подтверждалось экспериментально) бессимптомного.
Отмеченная особенность, т.е. неспособность вызывать острую форму инфекционного процесса, по-видимому, обусловлена особенностями самих возбудителей прионных болезней, так как уже давно обнаружено, что сам процесс накопления инфекционного прионного белка PrPSc в различных органах и тканях экспериментально зараженного лабораторного животного протекает весьма медленно. Можно полагать, что низкая скорость накопления инфекционного агента в данном случае обусловлена событиями, лежащими в основе механизма превращения клеточного прионного белка (PrPC ) в инфекционный прионный белок (PrPSc ).
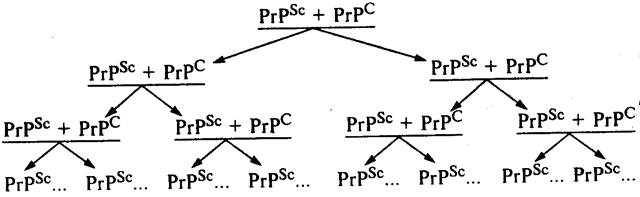
Собственно механизм накопления инфекционного белка в зараженном организме сегодня точно неизвестен. Вместе с тем, исходя из определения, что это посттрансляционный процесс, очевидно, что инфекционный прионный белок вызывает в здоровом организме трансформацию нормального прионного белка в инфекционную форму за счет его (нормального белка) конформационных (т.е. пространственных) изменений. В этом случае речь идет об изменении третичной или даже четвертичной структуры исходного белка PrPC . Таким образом, процесс накопления инфекционного прионного белка происходит не в результате синтеза в зараженном организме молекул PrPSc denovo, а вследствие конформационных изменений уже синтезированных перед этим нормальных молекул PrPC под влиянием инфекционного прионного белка PrPSc (схема). Процесс накопления инфекционного прионного белка обусловлен прежде всего необходимостью контакта двух молекул. В результате под влиянием одной молекулы PrPSc происходит трансформация одной молекулы PrPC в ее инфекционную форму PrPSc . Следующий этап, как видно на схеме, включает в себя уже наличие влияния двух молекул PrPSc , под воздействием которых образуются уже четыре молекулы PrPSc и т.д. Таким образом, как видно из приведенной схемы, процесс накопления инфекционного прионного белка носит лавинообразный характер.
Процесс накопления молекул инфекционного прионного белка
СТРУКТУРА ПРИОННЫХ БЕЛКОВ
Установленные необычные свойства возбудителей ТГЭ послужили основанием для выдвижения большого количества разнообразных теорий, пытающихся объяснить структуру и химическую природу этих агентов, многие из которых теперь имеют лишь историческое значение. Резкий скачок вперед в понимании природы возбудителей ТГЭ был сделан в результате разработок эффективных методов очистки и концентрации агента скрепи. Существенный вклад в разработку таких методов внесла группа Стенли Прузинера из Калифорнийского университета (США). Разработанная им многоступенчатая система очистки позволила получить препараты, очищенные в 100 – 1000 раз. На основании изучения высокоочищенных препаратов авторы пришли к выводу о том, что возбудитель скрепи является белком. Этот вывод был сделан в результате анализа инактивации агента при его обработке протеазой К, модификации при воздействии диэтилпирокарбонатом, додецилсульфатом натрия, гуанидинтиоцианатом, фенолом и мочевиной. Агент оставался устойчивым к обработке рядом реагентов, инактивирующих нуклеиновые кислоты, что указывало на их отсутствие в его составе. Изучение очищенного препарата возбудителя скрепи показало, что он обладает молекулярной массой около или меньше 50 000 Да.
Следует отметить, что представление о прионной природе возбудителя скрепи, выдвинутое С.Прузинером, оказалось очень плодотворным и послужило основанием для более детального распознавания природы возбудителей ТГЭ. В результате дальнейшей очистки приона было показано, что его основным компонентом является мажорный белок с молекулярной массой 27000 – 30000 Да, обозначаемый как РrР 27–30. Этот белок является составной частью скрепи-ассоциированных фибрилл, причем получены структурные и биохимические свидетельства того, что сборка этих фибрилл происходит invivo, и изучены некоторые молекулярные механизмы их образования. По своей физико-химической характеристике РrР представляет собой сиалогликопротеин и является первым идентифицированным структурным компонентом приона скрепи. Появление РrР 27–30 на этапе развития инфекции до развития гистопатологических изменений указывало на то, что этот белок не является вторичным продуктом патологической реакции. Был сделан вывод о том, что РrР 27–30 играет центральную роль в патогенезе скрепи.
При дальнейшем изучении прионов, выделенных из головного мозга зараженных скрепи животных, было выявлено наличие в ЦНС частиц в виде стержней диаметром 10 – 20 нм и длиной 100 – 200 нм. Ультраструктурно они напоминали амилоид и, по-видимому, представляли собой полимерную форму приона скрепи; каждый стержень содержал около 1000 молекул приона. Был проанализирован аминокислотный состав PrP 27–30 и определена последовательность 15 аминокислотных остатков в его полипептидной цепи. В последующем из головного мозга зараженных скрепи хомяков был выделен мажорный белок с молекулярной массой 33–37 кДа, обозначенный как HaSp 33–37; его выделение проводилось без этапа обработки протеазами. Обработка HaSp 33–37 протеазой К приводила к получению продукта, электрофоретически неотличимого от РrР 27–30. Была определена последовательность 22 аминокислотных остатков HaSp 33–37. Авторы полагали, что HaSp 33–37 представляет собой интактную форму белка возбудителя скрепи. Были изучены также некоторые другие характеристики прионов скрепи и болезни Крейтцфельдта–Якоба. В частности, при изучении липосом было подтверждено предположение о том, что инфекционная частица скрепи содержит 2 молекулы PrPSc и показано наличие вставок в ген приона при семейных случаях болезни Крейтцфельдта–Якоба и синдрома Герстманна–Штреусслера–Шейнкера.
Важным шагом, имеющим как теоретическое, так и методическое значение, было получение антител при использовании в качестве антигенов высокоочищенных прионов скрепи. В сыворотках кроликов, иммунизированных РrР 27–30, определяли антитела, специфически реагирующие с РrР 27–30 и с несколькими белками с более низкой молекулярной массой, очевидно, имеющими общую антигенную детерминанту с РrР 27–30 или являющимися продуктами его расщепления. Полученные антисыворотки не взаимодействовали с соответствующими белками, выделенными из головного мозга нормальных незараженных животных. Используя полученную антисыворотку с пероксидазной меткой, удалось показать локализацию прионов в определенных отделах головного мозга зараженных животных. В соответствии с ранее полученными данными структуры, связанные с меченой антисывороткой, обладали характеристикой амилоидных бляшек. Получение и использование антисыворотки к синтетическому пептиду, соответствующему N-концевой части приона скрепи, позволили провести индикацию белка скрепи-ассоциированных фибрилл в головном мозге, селезенке и лимфатических узлах зараженных животных. При этом положительные результаты были получены на ранних этапах инкубационного периода скрепи.
Развитие представлений о прионной природе возбудителя скрепи позволило сделать еще один решающий шаг в познании природы этих необычных агентов. В 1985 г. группе исследователей удалось выделить и охарактеризовать ген, кодирующий PrP 27–30. Оказалось, что этот ген содержится в ДНК, выделенной из мозга как скрепи-инфицированных, так и нормальных животных; соответственно м РНК для PrPC была обнаружена в головном мозге и в других тканях как инфицированных скрепи, так и нормальных животных. Используя соответствующую антисыворотку, удалось показать, что в тканях незараженных животных содержится белок, антигенно родственный PrP 27–30, но отличающийся от него чувствительностью к обработке протеазой К. Были получены доказательства того, что PrPC не кодируется гипотетической нуклеиновой кислотой агента. Эта точка зрения поддерживается в работах R.M.Ridley, H.F.Barker (1997). На основе этих данных были изучены биогенез и трансмембранная ориентация клеточной изоформы белка приона скрепи.
ФИЗИКО-ХИМИЧЕСКИЕ СВОЙСТВА ПРИОНОВ
Физико-химические свойства прионных белков особенно интенсивно изучались в последние годы, в результате чего были сформированы представления и получены новые данные о первичной, вторичной и третичной структуре PrP . Так, при анализе первичной структуры PrPC различных видов животных было выявлено, что 80% последовательностей PrPC у разных видов животных были идентичными. Исключение составлял куриный PrPC , где идентичность последовательностей по отношению к другим видам составляла всего 30%. Тем не менее 24 аминокислотные последовательности, располагающиеся между 112-м и 135-м аминокислотными остатками, являются высококонсервативными для всех видов млекопитающих, а также кур. В частности, было показано, что конверсия нормального прионного белка PrPC в его инфекционную форму (PrPSc ) является посттрансляционным процессом. Анализ вторичной структуры PrPSc выявил, что этот переход характеризуется большими структурными изменениями самого приона. Продемонстрировано, что PrPC содержит 42% a-спиралей и почти не содержит b-тяжей (около 3%), в то время как в его инфекционной форме PrPSc выявляется 30% a-спиралей и 43% b-тяжей. В экспериментальных исследованиях было подтверждено, что обработка нормального PrPC реагентами, уменьшающими образование b-тяжей, также приводит к уменьшению инфекционности приона; одновременно снижается и устойчивость к действию протеазы К, чувствительность к которой является маркером, отличающим PrPC от PrPSc .
Проведенный сравнительный анализ показал, что конформационные различия между нормальным и инфекционным прионным белком заключаются в трехмерной конформации. Переход нормального PrPC в его патологическую форму имеет в своей основе перестройку укладки белка. Корреляция изменений во вторичной структуре PrP с изменениями его инфекционности вместе с изменением конформации PrP дает основания заключить, что конформация прионного белка может иметь главное значение для проявления его патогенных свойств.
Были изучены некоторые закономерности перехода клеточной формы приона PrPC в его инфекционную форму и выявлено, что эффективность этой конверсии определяется видовой гомологией PrPC и PrPSc и, следовательно, в условиях гетерологичности обеих форм прионного белка эффективность конверсии снижается. Этим и объясняется механизм низкой инфекционности прионов в гетерологичной системе (например, животные – человек). Значение этой конверсии в развитии инфекционного процесса было подчеркнуто в экспериментальных исследованиях, показавших, что мыши, не экспрессирующие PrPC , устойчивы к инфекции прионами.
В исследованиях invitro, проведенных на модели агентов ТГЭ, также была установлена корреляция между эффективностью конверсии PrPC в PrPSc и способностью к трансмиссии агентов скрепи, губкообразной энцефалопатии коров и болезни Крейтцфельдта – Якоба. Было продемонстрировано, что конверсия PrPC в PrPSc в гетерологичной системе значительно снижена по сравнению с гомологичной системой. Авторы делают из своей сугубо экспериментальной работы практически важный вывод о том, что способность агентов скрепи и губкообразной энцефалопатии коров поражать людей после соответствующей экспозиции является ограниченной и низкой.
Таким образом, в результате разносторонних исследований, особенно интенсивно проводившихся в 90-е годы, были получены и систематизированы имеющие принципиальное значение данные о структуре и физико-химических свойствах прионных белков. Получение и анализ этих сведений создали необходимые предпосылки для дальнейшего углубленного изучения биологических особенностей прионных белков и механизма развития вызываемых ими заболеваний людей и животных.
БИОЛОГИЧЕСКИЕ ОСОБЕННОСТИ ПРИОНОВ
В последние годы вопрос о биологическом значении PrPC был подвергнут ревизии. На мышах, гомозиготных по потере гена PrPC , было показано, что эти животные после рождения росли нормальными, но спустя 70 нед у них развивались прогрессирующие симптомы атаксии, нарушалась моторная координация и отмечалась экстенсивная потеря клеток Пуркинье. Авторы сделали вывод о том, что PrPC играет важную роль в выживании клеток Пуркинье. Помимо этого, указывается на роль PrPC в регуляции циркадианных ритмов, на возможное участие PrPC в активации лимфоцитов и на его роль в качестве трофического фактора для некоторых популяций нейронов. Сохранность PrPC имеет значение для реализации нормальной функции синапсов. В последние годы опубликованы данные, свидетельствующие о роли PrPC в регуляции сна и продемонстрировано значение нарушения нормальной функции PrPC в возникновении смертельной семейной бессонницы. В исследованиях invitro было также показано, что PrPC вовлекается в процессы регуляции содержания внутриклеточного Са2+ в нейронах, Интенсивные исследования биологической роли PrPC позволили прийти к заключению о значении нормального приона PrPC в сохранении резистентности нейронов и астроцитов к окислительному стрессу.
Таким образом, за последние годы были значительно расширены представления о биологической значимости PrPC . Было установлено, что PrPC синтезируется в эндоплазматической сети и довольно быстро деградирует: продолжительность его полураспада составляет всего 5 – 6 ч. Синтезированный PrPC , проходя через аппарат Гольджи, транспортируется на поверхность клетки, где он связывается с гликофосфатидилинозитолом. Синтезированный PrPC в дальнейшем переносится вдоль аксона при помощи быстрого антероградного транспорта. В отличие от PrPC инфекционный прионный белок PrPC первично аккумулируется в клетках, накапливаясь в цитоплазменных везикулах. Дальнейшее накопление PrPSc в синаптических структурах и связанная с этим дезорганизация синапсов, возможно, являются причиной развития глубоких неврологических дефектов и деменции.
Напомним, что заболевания, вызываемые прионами, характеризуются рядом признаков, сочетание которых определяется биологическими особенностями их возбудителей, это прежде всего длительный инкубационный период (от месяцев до десятков лет), отсутствие воспалительных изменений, хронически прогрессирующая патология, отсутствие ремиссий и выздоровления. Для прионных болезней характерен ряд отрицательных признаков, которые не наблюдаются при заболеваниях, вызываемых вирусами. К ним относятся отсутствие продукции интерферона и нечувствительность к интерферону, отсутствие в составе возбудителя инфекционных нуклеиновых кислот и неспособность прионов интерферировать с вирусами. Для прионных болезней характерны нечувствительность к иммуносупрессирующему или иммунопотенцирующему действию АКТГ, кортизона, циклофосфамида, g-лучей, антилимфоцитарной сыворотки, тимэктомии и спленэктомии, отсутствие влияния адъювантов. Для прионных болезней характерна также интактность В- и Т-клеток. Комбинация всех перечисленных признаков, каждый из которых не является чем-то уникальным, и определяет своеобразие прионных болезней.
Получение новых данных в отношении биологических особенностей прионов позволило заключить, что прионные болезни являются нейродегенеративными заболеваниями, в возникновении которых фундаментальную роль играют конформационные изменения, а сам механизм развития болезни является беспрецедентным.
Результаты проведенных исследований позволили с новых позиций подойти к вопросу о природе агентов ТГЭ, а вся сумма полученных новых знаний о прионах послужила основанием для оптимистического высказывания С.Прузинера о том, что "эра черного ящика биологии скрепи и болезни Крейтцфельдта – Якоба, возможно, подходит к концу". В 1997 г. за свои многолетние исследования медленных инфекций, вызываемых прионами, американский биохимик С.Прузинер был удостоен Нобелевской премии по биологии и медицине. Таким образом, мы встречаемся с не очень частым случаем, когда Нобелевская премия присуждается дважды на протяжении 20 лет за исследование одной и той же проблемы, что, безусловно, свидетельствует о значимости самой проблемы и о темпах ее изучения.
ПРИОННЫЙ ГЕН
Современный этап в исследовании молекулярных основ прионных заболеваний человека и животных связан с идентификацией гена, кодирующего прионный белок. Расшифровка аминокислотной последовательности этого белка, позволила выяснить структуру кодирующей области соответствующего гена. Этот ген, получивший название PRNP, был вскоре выделен и исследован в лаборатории Ч.Вэйссманна. Выделение гена PRNP позволило использовать для исследования этиологии и патогенеза прионных заболеваний весь арсенал современных методов молекулярно-генетического анализа. В настоящее время структура белка PrP и соответствующего гена известна для многих организмов. Ген PRNP оказался эволюционно-консервативным: он найден у многих млекопитающих и птиц. В структурном отношении гены PRNP млекопитающих тоже схожи: в генах всех млекопитающих область, кодирующая PrP , локализована только в одном экзоне (экзоны - области гена, представленные в структуре зрелой иРНК). У человека этот ген локализован на хромосоме 20, у мышей - на хромосоме 2. Ген PRNP присутствует и экспрессируется не только у больных, но и у здоровых животных. При этом, несмотря на то, что иРНК гена PRNP в тканях мозга взрослых животных экспрессируется конститутивно, у молодых животных ее количество меняется с возрастом. Наибольшее количество иРНК PRNP зарегистрировано в нейронах.
Примерно 10% всех прионных заболеваний человека относятся к так называемым семейным формам или болезням с наследственной предрасположенностью. Идентификация прионного гена позволила связать семейные формы этих заболеваний с конкретными мутациями в гене PRNP. Так, например, мутация, вызывающая замену пролина на лейцин в 102-м положении PrP оказалась связана с развитием синдрома Герстманна-Штреусслера-Шейнкера. Интересно, что эта мутация приводит к заболеванию не только людей, но и мышей. Мутация в 178-м кодоне может быть связана как с развитием болезни Крейтцфельдта-Якоба (БКЯ), так и смертельной семейной бессонницы. Интересно, что оба случая связаны с заменой аспарагиновой кислоты на аспарагин. Разница, по всей видимости, заключается в том, что мутантная аллель при смертельной семейной бессоннице в 129-м положении несет кодон для метионина, в то время как в случае БКЯ это положение занимает кодон для валина. Наследственная предрасположенность к прионным заболеваниям может быть связана не только с определенными аминокислотными заменами в PrP , но и с его гораздо более существенными изменениями. Так, в аминоконцевом районе PrP имеется 5 расположенных подряд идентичных последовательностей из 8 аминокислот. Некоторые формы семейной БКЯ оказались связанными с увеличением количества таких повторов. Механизм образования этих повторов неясен. Однако понятно, что в отличие от случаев, описанных ранее, они возникли не за счет точковых мутаций, а в результате рекомбинационных событий. Всего в настоящее время в гене PRNP человека известно около 20 мутаций, связанных с семейными формами прионных заболеваний.
У животных аллельный полиморфизм по гену PRNP тоже связан с вероятностью развития прионных болезней. Тем не менее вопрос о том, влияют ли изменения в структуре этого гена на вероятность спонтанного заболевания животных или они связаны с подверженностью животных прионным инфекциям, остается открытым.
Важной особенностью прионов как инфекционных агентов является наличие межвидовых барьеров на пути их передачи, хотя PrP лишь незначительно отличается по первичной структуре у разных видов млекопитающих. В большинстве случаев эти барьеры не абсолютны, иными словами, они не препятствуют, а лишь значительно затрудняют передачу инфекции от особей одного вида особям другого вида. Вместе с тем известно по крайней мере одно исключение из этого правила: у кроликов не удается вызвать заболевание после заражения их инфекционным прионным белком, выделенным из мозга самых разных животных. Хотя причина устойчивости кроликов к прионной инфекции точно неизвестна, анализ структуры гена, кодирующего PrP кролика, показал, что она может быть связана с некоторыми особенностями первичной структуры этого белка.
Существенный прогресс в исследовании прионов стал возможным после передачи скрепи мышам и хомякам. Как уже было упомянуто, делеция гена PRNP не приводит к немедленной смерти животных. Это позволило установить, что особи, лишенные гена PRNP, не заражаются прионами и, вероятно, вообще не подвержены этим заболеваниям. Интересно, что в подобных экспериментах было показано, что удаление аминоконцевой последовательности PrP мыши за счет делеции соответствующей области гена PRNP не препятствует развитию прионных заболеваний. Ранее было отмечено, что некоторые семейные формы БКЯ связаны с увеличением количества аминокислотных повторов в этой области белка PrP человека. Таким образом, эти изменения влияют на вероятность заболевания, хотя его наличие не является критичным для прионного превращения белка.
Увеличение экспрессии гена PRNP (количество PrP ) способствует возникновению заболевания. В силу существования межвидовых барьеров мыши устойчивы к заражению прионами, выделенными из мозга больных хомяков, но трансгенных мышей, несущих ген PRNP хомяка, легко заразить с помощью инокуляции суспензии клеток мозга больного хомяка. Все перечисленные факты полностью согласуются с теорией о белковой природе прионов. Животные, лишенные гена PRNP, не заболевают просто потому, что их клетки не содержат белка, подверженного конформационной перестройке. При увеличении количества молекул этого белка должна возрастать вероятность спонтанного перехода какой-либо из молекул в патогенную форму. Существование наследственной предрасположенности к прионным заболеваниям связано с тем, что мутации увеличивают вероятность прионного превращения белка PrP . Менее выраженная способность к инфицированию у "чужого" приона при межвидовом заражении может объясняться пониженной способностью PrPSc передавать свое патогенное состояние PrPC несколько отличающемуся от него по первичной структуре.
Итак, возможность заражения особей одного вида с помощью прионов, выделенных из тканей мозга особей другого вида, способствовала использованию лабораторных животных (мышей и хомяков) для изучения природы прионов и вызываемых ими болезней. В то же время отсутствие межвидовых барьеров на пути распространения прионов означает принципиальную возможность их передачи от животных человеку. Действительно, в последнее время проблема прионов приобрела существенное практическое значение в связи со вспышками соответствующих эпизоотии среди сельскохозяйственных животных в некоторых европейских странах, а также с появлением наблюдений о возможности передачи этих заболеваний от животных человеку. В настоящее время получен ряд серьезных свидетельств, указывающих на опасность заражения человека прионами животных. При этом наиболее убедительные доказательства базируются на результатах, полученных с использованием трансгенных лабораторных животных.
ПАТОЛОГИЯ ЦЕНТРАЛЬНОЙ НЕРВНОЙ СИСТЕМЫ
ПРИ ПРИОННЫХ БОЛЕЗНЯХ ЧЕЛОВЕКА
В настоящее время известно 4 прионных заболевания: болезнь Крейтцфельдта- Якоба (БКЯ), куру, синдром Герстманна-Штреусслера-Шейнкера (СГШШ) и фатальная семейная инсомния (ФСИ). Основную массу прионных болезней составляет БКЯ, чаще всего в виде спорадических случаев, в 10% случаев БКЯ носит семейный характер. Наблюдается также ятрогенная форма БКЯ, которая, как и куру, манифестирует как инфекция в результате случайного заражения прионными болезнями. СГШШ и ФСИ являются доминантно наследуемыми прионными болезнями, которые, как было показано, вызываются мутациями прионного гена.
В нашей стране опубликованы лишь единичные работы с описанием морфологических изменений ЦНС при спорадических случаях БКЯ. Нами опубликованы данные о прижизненной морфологической диагностике двух спорадических случаев БКЯ на основе исследования биоптатов коры большого мозга с использованием световой и электронной микроскопии, в которых диагноз в дальнейшем был подтвержден на аутопсии. Отсутствуют работы, посвященные морфологическим изменениям мозга при других формах прионных заболеваний. В то же время за рубежом в последние годы значительно возросло число публикаций, в том числе и обобщающих, в которых на основании уже довольно большого числа наблюдений подробно описаны особенности изменений ЦНС при всех на сегодняшний день известных формах прионных заболеваний, включая куру, БКЯ (спорадическую, наследственную, ятрогенную формы и новый вариант), СГШШ и ФСИ. Помимо морфологических исследований с использованием классических нейрогистологических методик, эти работы включают в себя и данные иммуноцитохимического исследования, направленные на выявление отложений патологической изоформы прионного белка (PrPSc ) в гистологических срезах из различных областей мозга. Без преувеличения можно сказать, что именно эти методы, направленные на идентификацию отложений PrPSc в ткани мозга, "революционизировали" прижизненную или посмертную диагностику прионных заболеваний, позволяя поставить уверенно достоверный диагноз в ранних стадиях заболевания, в том числе и до развития в мозге характерных морфологических изменений. В последние годы под эгидой ВОЗ разработаны критерии морфологической диагностики этих заболеваний. Учитывая особую эпидемиологическую значимость и связь заболевания со спонгиоформной энцефалопатией крупного рогатого скота, основной акцент сделан на разработку критериев морфологической диагностики нового варианта БКЯ.
При морфологическом исследовании мозга больных, погибших от различных прионных болезней, выявлены черты их сходства и различия. Макроскопически выявляется снижение объема и массы головного мозга и уменьшение толщины (атрофия) его коры. Степень выраженности этих изменений тесно связана с продолжительностью жизни больных, однако может и не выявляться каких-либо макроскопических изменений мозга. Хотя атрофия коры мозга является характерной находкой во многих случаях БКЯ, выраженность ее широко варьирует в пределах различных областей коры, в разных случаях. Характер корковой атрофии может быть связан с клиническими проявлениями заболевания. Так, в случаях корковой слепоты выявляется выраженная атрофия коры затылочных долей мозга.
Изредка масса мозга при БКЯ значительно уменьшена (менее чем 1000 г), атрофия коры в таких случаях обычно сопровождается атрофией базальных ядер, таламуса и гипоталамуса. Избирательная атрофия таламуса характерна для ФСИ и может быть выявлена при макроскопическом исследовании мозга. Эти макроскопические изменения неспецифичны и могут наблюдаться при широком круге других нейродегенеративных заболеваний, включая болезнь Альцгеймера, Пика, хорею Гентингтона и мультисистемную атрофию. Атрофия мозжечка может быть макроскопически ярко выражена при некоторых прионных заболеваниях человека, особенно при куру и СГШШ, a также при ятрогенной БКЯ, развившейся у больных, которым проводилось лечение человеческим гормоном роста. В этих случаях атрофия коры большого мозга может отсутствовать. Дифференциальная диагностика должна проводиться в первую очередь с различными формами спиноцеребеллярной дегенерации.
Для прионных болезней человека характерны следующие общие гистологические изменения: спонгиоформная дегенерация серого вещества головного мозга, атрофия и гибель нервных клеток, астроцитарный глиоз, амилоидные бляшки, содержащие PrPSc . При различных формах прионных заболеваний эти изменения непостоянно присутствуют во всех отделах ЦНС и широко варьируют от случая к случаю и в пределах ЦНС в отдельных случаях. Так, при БКЯ указанные изменения регистрируются в коре большого мозга, базальных ядрах, таламусе, молекулярном слое коры мозжечка и верхней части ствола мозга, причем амилоидные бляшки в спорадических случаях обнаруживаются в 5 - 10% случаев.
Все авторы подчеркивают, что существует широкий спектр морфологических изменений ЦНС (спонгиоз, гибель нейронов, астроцитоз и амилоидные бляшки) при прионных заболеваниях как в отношении их распространенности и тяжести, так и в отношении выявления и локализации в мозге. Оказалось, что при некоторых прионных заболеваниях, в частности при ФСИ, отсутствует такой кардинальный морфологический признак прионных заболеваний, как спонгиоформная дегенерация серого вещества, а недавно появились сообщения о наследственных прионных заболеваниях (так называемые атипичные прионные деменции), при которых отсутствуют все классические морфологические проявления БКЯ.
Все это потребовало унификации патоморфологических критериев диагностики БКЯ и других прионных заболеваний человека, что и сделано в настоящее время под эгидой ВОЗ. Это представляется особенно важным, так как результаты нейроморфологического исследования играют ключевую роль в постановке достоверного диагноза БКЯ, при этом, кроме биопсии мозга, до сих пор не существует других специфических методов прижизненной диагностики БКЯ. Ведущее значение имеет выявление иммуноцитохимическими методами отложений PrPSc в ткани мозга.
Болезнь Крейтцфельдта-Якоба. БКЯ является наиболее распространенным нейродегенеративным заболеванием прионной природы, составляющим от 88 до 90% всех прионных болезней человека. Основная масса наблюдений приходится на спорадические случаи БКЯ, в которых отсутствует связь с приобретенной инфекцией или наследственностью. Около 10% семейных случаев БКЯ обусловлены врожденными мутациями в гене PRNP. Выделяется также имеющая свои клинико-морфологические особенности ятрогенная форма БКЯ, связанная с заражением пациентов при проведении им операций с использованием инструментария, который использовался ранее при операциях у больных с недиагностированной БКЯ. В последние годы за рубежом (Великобритания, Франция) идентифицирован новый вариант БКЯ, этиологически, как это установлено, связанный с прионами, вызывающими развитие губкообразной (спонгиоформной) энцефалопатии крупного рогатого скота (ГЭКРС), и характеризующийся патоморфологическими изменениями мозга, отличными от классического варианта БКЯ.
Спорадическая БКЯ. Макроскопические изменения мозга при спорадической БКЯ отличаются значительной вариабельностью. В некоторых случаях никаких отчетливых изменений не выявляется, в то время как в других обнаруживается различная степень атрофии коры мозга, базальных ядер и мозжечка со снижением массы мозга до 850 г. При макроскопическом исследовании головного мозга умерших с развернутой картиной БКЯ нами выявлено выраженное уменьшение массы мозга и резкое истончение коры, преимущественно коры лобных долей и предцентральных извилин со значительным расширением борозд данных областей. На горизонтальных разрезах головного мозга по Флексигу на уровне базальных ядер и таламуса обнаруживается некоторое снижение их объема, расширение желудочков мозга, а также значительное уменьшение толщины коры мозга в области всех долей: лобной, теменной, затылочной и височной. Следует подчеркнуть, что такие выраженные явления уменьшения массы и объема мозга у больных с БКЯ могут выявляться с помощью компьютерной томографии в виде расширения борозд и желудочков.
Классическая триада гистологических изменений мозга при БКЯ складывается из спонгиоформной дегенерации нейронов и их отростков, гибели нейронов и интенсивного реактивного астроцитоза, однако выраженность и распространенность каждого из этих феноменов может значительно варьировать. Что касается 4-го морфологического феномена - формирования амилоидных бляшек, то в классических спорадических случаях он наблюдается редко.
Следует отметить, что указанная триада морфологических изменений мозга (гибель нейронов, астроглиоз и спонгиоз) обнаруживается не только в различных отделах коры большого мозга, но и в базальных ядрах, таламусе и мозжечке. В мозжечке выраженные изменения обнаруживаются в виде значительной гибели нейронов зернистого слоя, сморщивания и гиперхроматоза клеток Пуркинье, пролиферации астроцитов. В качестве характерной патологии отдельных клеток Пуркинье при БКЯ и других прионных заболеваниях отмечается локальное набухание их аксонов - "торпеды". Отмечается спонгиоз I (молекулярного) слоя, хотя сливающиеся вакуоли нехарактерны для этой области.
Вакуолизация белого вещества полушарий большого мозга не всегда (в отличие от серого вещества) выявляется при БКЯ. В основном изменения белого вещества, сопровождающиеся уменьшением числа миелиновых волокон, фрагментацией и набуханием аксонов, обнаруживаемые в отдельных случаях при БКЯ и других прионных заболеваниях, рассматриваются как вторичные. Однако в некоторых случаях БКЯ, в частности в случаях из Японии, обнаружены некротические изменения белого вещества с развитием вакуолярной миелопатии.
По характеру распространения основных характерных для БКЯ изменений в таких случаях можно говорить о панэнцефалопатической форме болезни, при которой эти изменения развиваются не только в коре различных областей полушарий большого мозга, базальных ядрах, таламусе и мозжечке, но и в белом веществе полушарий мозга. В литературе описан подобный панэнцефалопатический вариант БКЯ. Развивающуюся при нем дегенерацию белого вещества, обозначаемую как вакуолярная миелопатия, авторы рассматривают как самостоятельный патологический процесс, а не как вторичные изменения в результате гибели нервных клеток.
Следует особо подчеркнуть, что в работах последних лет морфологическая картина БКЯ была дополнена еще одним элементом, а именно куру-бляшками или подобными им амилоидными бляшками, представляющими собой амилоидные агрегаты сферической формы с радиальной исчерченностью по периферии, в которых содержится PrPSc , выявляемый с помощью иммуноцитохимических методов, которые в настоящее время являются наиболее чувствительными и специфическими методами обнаружения бляшек. Впервые амилоидные бляшки, имеющие подобную структуру, были описаны при куру, откуда и получили свое название "куру-бляшки", используемое для обозначения подобных бляшек, встречающихся и при других прионных заболеваниях. В отличие от куру, при которой бляшки выявляются в 70% случаев, в спорадических случаях БКЯ куру-бляшки обнаруживаются только в 5-10% случаев. Имеются данные о том, что их наличие взаимосвязано с более длительным клиническим течением заболевания. Выявление куру-бляшек служит важным диагностическим критерием для постановки достоверного диагноза БКЯ.
Классические компактные бляшки при БКЯ и куру состоят из гомогенного плотного центра, окруженного бледным ореолом радиальных фибрилл. По данным литературы, при куру и БКЯ они с наибольшей частотой выявляются в зернистом слое коры мозжечка, но иммуноцитохимическое исследование позволяет выявить более широкое их распространение в мозжечке, включая молекулярный слой коры и подлежащее белое вещество. Реже сходные бляшки могут быть выявлены в других отделах мозга, включая кору, базальные ядра и таламус. Установлено, что при отсутствии бляшек куру в мозжечке они не выявляются и в большом мозге. Формирование этих бляшек наиболее характерно для описанных в последнее время случаев нового варианта БКЯ, а также ятрогенной БКЯ. Многочисленные куру-бляшки наблюдаются и при таком прионном заболевании, как СГШШ, для которого особенно характерна локализация таких бляшек в мозжечке.
На основании иммуноцитохимического исследования мозга при прионных заболеваниях установлено, что, помимо амилоидных бляшек, PrPSc может откладываться в спонгиоформно измененной коре полушарий большого мозга в виде гранул, окружающих вакуоли, что, как полагают, соответствует аккумуляции его в синапсах. В коре мозжечка такие отложения PrPSc локализуются в зернистом слое между телами нейронов. Менее значительные гранулярные скопления PrPSc могут наблюдаться по ходу проводящих путей в мозговом стволе, в синапсах на поверхности нейронов, иногда и интранейронально. В случаях выраженного глиоза в коре большого мозга и базальных ядрах выявлено гранулярное иммуноокрашивание цитоплазмы астроцитов аналогично тому, как это наблюдалось у животных.
Новый вариант БКЯ. В последние годы наряду с классическим вариантом БКЯ более детально были изучены патологические изменения ЦНС при других прионных заболеваниях. Это касается в первую очередь нового варианта БКЯ, возникновение которого связывают с передачей прионов от больных коров, а также СГШШ и ФСИ.
Патоморфологические особенности нового варианта БКЯ отражены в рекомендациях ВОЗ по диагностике трансмиссивных спонгиоформных энцефалопатий человека. В них подчеркивается, что новый вариант БКЯ включает в себя основные патологические процессы, характерные для всех трансмиссивных спонгиоформных энцефалопатий человека: спонгиоформные изменения, утрату нейронов, реактивный астроцитоз и накопление в мозге изоформы PrPSc , связанной с заболеванием. Однако природа и локализация этих изменений при новом варианте (в отличие от спорадической БКЯ) являются относительно постоянными и отличаются от таковых при других формах спонгиоформной энцефалопатий человека. Основные проявления могут быть суммированы следующим образом.
Для нового варианта БКЯ характерны множественные бляшки, имеющие фибриллярную структуру, содержащие PrPSc и локализующиеся в коре полушарий большого мозга и мозжечка, часто окруженные ореолом спонгиоформных изменений - "цветущие" бляшки. Подобные бляшки встречаются во всех отделах мозга и мозжечка. Хотя крупные бляшки визуализируются на препаратах, окрашенных гематоксилином и эозином, неоценимым методом диагностики является иммуноцитохимическое исследование на выявление PrPSc . Только с помощью этого метода, помимо "цветущих" бляшек, выявляются множественные мелкие бляшки, содержащие PrPSc , которые образуют скопления в коре полушарий большого мозга и мозжечка и не связаны со спонгиоформными изменениями, а также аморфные отложения PrPSc вокруг нейронов и сосудов в этих же областях коры большого мозга и мозжечка.
Полный спектр морфологических изменений, характерных для нового варианта БКЯ, также включает в себя спонгиоформные изменения, наиболее выраженные в базальных ядрах с массивным отложением PrPSc ; резко выраженный астроглиоз и гибель нейронов в таламусе, особенно в его дорсомедиальном и заднем ядрах; массивное накопление PrPSc , часто в виде фокальных отложений, в коре мозжечка, включая ее молекулярный слой и слой клеток-зерен с редкими бляшками в белом веществе; точечное отложение PrPSc в ядрах моста.
Ятрогенная БКЯ. В последнее время продолжает увеличиваться число факторов риска передачи прионов от человека к человеку и возникновения ятрогенной БКЯ, которая в отличие от спорадических случаев развивается у больных более молодого возраста. Случаи передачи БКЯ людям связаны с пересадкой роговицы, имплантацией в мозг зараженных электродов и хирургическими операциями, в которых использовались зараженные инструменты или аппараты. С 1988 г. описано 11 случаев БКЯ после пересадки твердой мозговой оболочки. Спорадическая БКЯ редко наблюдается у больных до 40 лет, поэтому развитие БКЯ у 55 больных в возрасте от 10 лет до 41 года, которым проводилась гормонотерапия (инъекции человеческого гормона роста), также указывает на ятрогенную природу заболевания. Все эти больные получали гормон каждые 2-4 дня в течение от 4 до 12 лет. Имеются также сообщения о 5 случаях БКЯ, развившейся у женщин, получавших человеческий гипофизарный гонадотропин.
В случаях ятрогенной БКЯ могут быть выявлены все описанные выше патоморфологические изменения мозга, характерные для прионных заболеваний, хотя локализация изменений варьирует от случая к случаю. Установлена зависимость морфологических изменений мозга и клинических проявлений заболевания от характера заразного материала и способа его введения. Так, при внутримозговом заражении вследствие нейрохирургических процедур и операций по пересадке твердой мозговой оболочки развивается классическая клиническая картина БКЯ с выраженными спонгиоформными изменениями в коре большого мозга. Однако в случаях, связанных с лечением человеческим гормоном гипофиза, выявляется особенно тяжелое поражение мозжечка.
Что касается спонгиоформных изменений и отложений PrPSc в коре большого мозга, то в значительной части наблюдений ятрогенной БКЯ (в отличие от спорадической формы) они носят очень ограниченный характер. Клинико-морфологические сопоставления показали, что у большинства больных, получавших человеческий гормон роста, заболевание по клиническому течению больше напоминало куру. Оно характеризовалось длительным инкубационным периодом, колебавшимся от 4 до 30 лет, постепенным нарастанием мозжечковой симптоматики. Деменция у некоторых больных развивалась только в терминальной стадии заболевания.
Семейная БКЯ. В 10% случаев БКЯ носит семейный характер и связана с мутациями в гене PRNP. Еще до открытия генетической патологии было очевидно, что семейные случаи БКЯ имеют клиническую картину, несколько отличающуюся от проявлений классической спорадической БКЯ. Заболевание начинается обычно в более молодом возрасте и характеризуется большей продолжительностью. Первые проявления часто связаны с мозжечковыми симптомами, и патоморфологическое исследование обычно подтверждает тяжелое поражение мозжечка. При этом прослеживается определенный параллелизм между степенью поражения мозжечка и большей продолжительностью течения болезни. Высказывается предположение, что распространение инфекционного агента в ЦНС в семейных случаях БКЯ происходит более медленно, чем в спорадических случаях, однако в настоящее время этот процесс остается недостаточно изученным.
Отложения PrPSc , как и спонгиоформные и другие изменения мозга, часто топографически довольно ограничены и носят локальный характер, что обнаруживается только при тщательном морфологическом исследовании. В ряде семейных случаев БКЯ изменения более выражены в базальных ядрах и мозжечке, где выявляются куру-бляшки, содержащие PrPSc , при отсутствии в других отделах мозга изменений, характерных для прионных заболеваний.
Куру. Этот вариант прионных заболеваний человека связан с традицией каннибализма в племени Форе в восточном нагорье Папуа Новой Гвинеи. Ведущим клиническим проявлением куру является прогрессирующая дисфункция мозжечка и поэтому неудивительно, что атрофия мозжечка, преимущественно червя, была единственным проявлением заболевания, обнаруженным при макроскопическом исследовании мозга в таких случаях.
Микроскопически наиболее выраженные изменения также локализуются в мозжечке. Они выражаются в утрате нейронов зернистого слоя, а также грушевидных нейронов (клетки Пуркинье), в веретенообразном набухании проксимальных отделов аксонов многих оставшихся клеток Пуркинье ("торпеды") и интенсивном глиозе с пролиферацией глии Бергмана.
Синдром Герстманна-Штреусслера-Шеинкера (СГШШ). СГШШ представляет собой доминантно наследуемую форму прионного заболевания, клинически проявляющуюся прогрессирующей спинно-мозжечковой атаксией с деменцией, развитие которой обусловлено мутацией в гене PRNP.
Наиболее характерным морфологическим признаком СГШШ является отложение мультицентрических амилоидных бляшек практически во всех отделах мозга, но наиболее многочисленны они в мозжечке, где располагаются преимущественно в молекулярном и зернистом слоях коры; в меньших количествах они обнаруживаются в белом веществе мозжечка, в коре полушарий большого мозга и базальных ядрах. Мультицентрические бляшки при СГШШ представлены крупными центрально расположенными массами амилоида, вокруг которых имеются более мелкие дополнительные отложения. Куру-подобные (моноцентрические) амилоидные бляшки, характерные для куру и спорадической БКЯ, также могут выявляться при СГШШ.
Наряду с многочисленными амилоидными бляшками для СГШШ характерны изменения мозжечка, что выявляется уже при его макроскопическом исследовании в виде выраженной атрофии. Микроскопически особенно характерна дегенерация зубчатого ядра, сочетающаяся с гибелью нейронов и распространенным астроглиозом.
Фатальная семейная инсомния (ФСИ). При ФСИ, представляющей собой доминантно наследуемую форму прионного заболевания, наиболее выраженные морфологические изменения развиваются в вентральном и дорсомедиальном ядрах таламуса, где погибает более 50% нейронов и развивается реактивный астроцитоз. Спонгиоформные изменения в указанных отделах таламуса также могут выявляться, но на первый план выступают дегенеративные изменения нейронов таламуса, выраженный астроглиоз в сочетании с отложением PrPSc при отсутствии спонгиоза. В других отделах таламуса, а также в коре большого мозга и мозжечка выпадения нейронов и глиоз значительно менее выражены. Изменения в коре большого мозга минимальны. Они выражаются в виде небольшой очаговой пролиферации астроцитарной глии. Лишь в одном случае были выявлены слабовыраженные спонгиоформные изменения, преимущественно во II и IV слоях коры. | В мозжечке отмечаются веретенообразное набухание аксонов на их отдельных участках ("торпеды") многих клеток Пуркинье и гибель небольшого числа клеток Пуркинье и клеток-зерен. Амилоидные бляшки отсутствуют. Отмечается утрата нейронов в нижней оливе более чем на 50%.
Установлено, что при ФСИ, несмотря на отсутствие выраженных морфологических изменений в различных областях мозга, иммуноцитохимически выявляется распространенное отложение патологической изоформы PrPSc , устойчивой к протеазе. Это согласуется с гипотезой, что накопление PrPSc предшествует развитию морфологических изменений мозга и является причиной нарушения функции нейронов.
***
Расширение клинического спектра прионных болезней человека произошло благодаря достижениям в области молекулярной генетики и нейрохимии прионных заболеваний. Эти достижения в свою очередь сопровождались более детальным изучением патогенетических механизмов и структурных изменений, развивающихся в ЦНС при различных формах этих заболеваний. Современные методы исследования, особенно иммуноцитохимические, позволили выявить более широкий спектр изменений в ЦНС, чем это было описано ранее, до открытия отложений PrPSc , в классических работах по патоморфологии прионных заболеваний. Эти данные важны при дифференциальной диагностике прионных болезней человека и других нейродегенеративных заболеваний. Оказалось, что спектр иммуноокрашивания, отражающий отложение PrPSc в ткани мозга, характеризуется такой же вариабельностью, как и спектр морфологических изменений, описанных в классических публикациях. Поэтому для установления закономерностей локализации PrPSc при разных прионных болезнях необходимо исследовать важнейшие отделы мозга - участки коры большого мозга из лобной, теменной, височной и затылочной долей, а также базальных ядер, таламуса, гипоталамуса, ствола мозга и мозжечка. Дальнейшие перспективы связывают с экспериментальным воспроизведением прионных заболеваний у трансгенных животных, что позволит детально исследовать эволюцию структурных изменений в ЦНС при различных формах прионных болезней, а также изучить взаимоотношения между морфологическими изменениями и отложениями PrPSc в ткани мозга. Таким образом, иммуноцитохимия стала важнейшим инструментом, расширяющим наше понимание основных патологических процессов, развивающихся в головном мозге при прионных заболеваниях человека. При установлении закономерностей изменений и локализации отложений PrPSc , возможно, будет найден ключ к пониманию патогенеза этого заболевания.
Значительное сходство патологической картины мозга при прионных заболеваниях человека и при болезни Альцгеймера указывает на существование общих механизмов, приводящих к изменениям нейронов и их гибели при этих неизлечимых заболеваниях. Оба заболевания чаще развиваются как спорадические формы, а в семейных случаях заболевание наследуется по аутосомно-доминантному типу и обусловлено специфическими генетическими мутациями. Утрата нейронов и реактивный глиоз отмечаются в ЦНС при обоих заболеваниях. Установлен общий механизм формирования и эволюции амилоидных бляшек в мозге при болезни Альцгеймера и прионных заболеваниях, в котором принимают участие отростки нейронов, микроглиальные клетки и отростки астроцитов. Однако между этими заболеваниями имеются и различия. В первую очередь это характер белка, входящего в состав амилоидных бляшек. Кроме того, изменения нейрофибрилл, являющиеся наиболее распространенной формой патологии нейронов при болезни Альцгеймера, очень редко наблюдаются при прионных заболеваниях. В противоположность прионным заболеваниям, при болезни Альцгеймера отсутствуют доказательства трансмиссивности. Все это свидетельствует о том, что перспективным направлением исследований прионных болезней человека является сопоставительный анализ изменений, развивающихся в ЦНС при других заболеваниях, относящихся к нейродегенеративным.
ИЗУЧЕНИЕ ПРИОННЫХ БОЛЕЗНЕЙ
В последние годы интерес к прионным болезням в мире резко возрос в связи с эпидемией ГЭКРС в Англии. В этот период, в основном в Англии, зарегистрировано 23 спорадических наблюдения нового варианта БКЯ, дебют которой отмечался в молодом возрасте, что нетипично для этого заболевания, причем при патогистологическом исследовании мозга умерших больных были выявлены изменения, сходные с таковыми при ГЭКРС. Это позволило высказать предположение о возможности заражения людей через продукты, производимые из мяса этих животных.
Ситуация еще больше обострилась в связи с тем, что в последние годы появились прецеденты прорыва существующих для прионов видовых барьеров. Так, прионные заболевания стали регистрироваться у животных, у которых в обычных условиях эта патология не наблюдается (кошки, а также некоторые животные, содержащиеся в неволе в зоопарке), что связывается с кормлением их продуктами, приготовленными из тканей животных - традиционных носителей этого инфекционного белка (овцы, козы).
И наконец, в самое последнее время появились сообщения об идентичности прионов, выделенных от больных с новым вариантом БКЯ и от коров с трансмиссивной спонгиоформной энцефалопатией. Эти работы были проведены в рамках Национального проекта наблюдения за БКЯ, который был создан в Великобритании в 1990 г. для оценки возможных влияний спонгиоформной энцефалопатии крупного рогатого скота на здоровье человека.
Приведенные данные свидетельствуют о реальной возможности заражения людей прионами при употреблении в пищу мясных продуктов, полученных из пораженных этими возбудителями животных.
До сих пор пересматривается терминология этих заболеваний. Термин "спонгиоформная энцефалопатия" неадекватен, поскольку, например, у больных с ФСИ этот признак выражен минимально. Термин "трансмиссивная энцефалопатия" также проблематичен из-за сложности демонстрации трансмиссивности. Термин "прионные болезни" наиболее приемлем, поскольку включает в себя уникальные особенности этих болезней и центральную роль прионов в их патогенезе, которая тем не менее может быть разной при спорадических, трансмиссивных и наследственных формах.
Прогресс в понимании природы прионных заболеваний человека связан с изучением возможности их передачи животным и с обнаружением прионного белка в результате молекулярного клонирования его гена PRNP, который картирован на коротком плече хромосомы 20, а также с созданием трансгенных мышей, экспрессирующих включенный в их геном (вместо собственного) мутировавший PrP ген. Патологическая изоформа прионного белка (PrPSc ) отличается от нормальной (PrPC ) своей высокой резистентностью к расщеплению протеазой К, нерастворимостью после очищающей экстракции, способностью накапливаться во вторичных лизосомах, посттрансляционным синтезом и обогащением во время процесса выделения прионного инфекционного начала. При этом PrPSc образуется путем конформации PrPC хозяина.
В настоящее время считается, что прионные заболевания возникают в результате накопления PrPSc , а не подавления функции PrPC . Первично PrPSc накапливается в клетке, где откладывается в цитоплазматических пузырьках, многие из которых являются вторичными лизосомами. В последующем этот протеин высвобождается во внеклеточное пространство и откладывается в амилоидных бляшках.
С углублением понимания этиологии классических прионных болезней человека (БКЯ, куру и СГШШ) в настоящее время считается целесообразным их разделение на спорадические, приобретенные и наследственные (тaбл.2). При этом они рассматриваются как клинико-патологические синдромы в рамках, по-видимому, более широкого спектра заболеваний. Свидетельством этому являются описание в последние годы не только фатальной семейной инсомнии, но и некоторых вариантов БКЯ и СГШШ, характеризующихся определенными мутациями гена прионного белка.
Таблица 2. Типы прионных болезней человека с учетом причины их возникновения |
||
Типы |
Клинический синдром |
Причина заболевания |
| Спорадические | БКЯ Атипичная БКЯ |
Возможно, соматическая мутация PRNP или спонтанная конверсия PrPC в PrPSc |
| Приобретенные(инфекционные) | Куру Ятрогенная БКЯ Новый вариант БКЯ |
Каннибализм Инокуляция Алиментарный путь |
| Наследственные | Наследственная БКЯ СГШШ ФСИ Различные атипичные деменции |
Мутации PRNP |
Так, описаны семьи с наследственными прионными заболеваниями, в которых отмечался выраженный фенотипический полиморфизм, напоминающие БКЯ и СГШШ, равно, как были случаи, не имеющие сходства с БКЯ и СГШШ. В литературе приведены наблюдения, идентифицированные путем анализа PRNP-гена, которые были атипичны не только клинически, но и морфологически, поскольку в них полностью отсутствовали классические патогистологические признаки прионных болезней. В отдельных семейных случаях БКЯ обращается внимание на их клиническое сходство с рядом нейродегенеративных заболеваний, такими как болезнь Альцгеймера, болезнь Пика, лобная дегенерация неальцгеймеровского типа, боковой амиотрофический склероз с деменцией. Следует отметить, что в последние годы в литературе сложилось мнение о целесообразности обозначения семейных случаев прионных болезней как наследственных с подразделением их в зависимости от характера мутации PRNP-гена. К настоящему времени обнаружено более 20 мутаций PRNP, достоверно связанных с врожденными прионными заболеваниями.
Высокий уровень разнообразия клинических проявлений предполагает возможность роли других генетических факторов в патогенезе прионных заболеваний. Одним из таких кандидатов в настоящее время является аполипопротеид Е (ароЕ), ген которого картирован на хромосоме. Очевидно, генотип ароЕ может влиять на начало заболевания и характер патоморфологических изменений при спорадической БКЯ. Так, показано, что позднее начало этой формы БКЯ связано с apoEe2. Однако это направление нуждается в более углубленном исследовании.
Приобретенные прионные болезни включают ятрогенную БКЯ, новый вариант БКЯ и куру. К спорадическим прионным болезням относят БКЯ и атипичные варианты БКЯ, последние - в связи с возможностью передачи приматам, даже при отсутствии характерных патогистологических признаков этого заболевания.
ЛЕЧЕНИЕ И ПРОФИЛАКТИКА ПРИОННЫХ БОЛЕЗНЕЙ ЧЕЛОВЕКА
В настоящее время не существует эффективной этиологической и патогенетической терапии прионных болезней, несмотря на достигнутый в последние годы прогресс в изучении этой группы медленных инфекций.
В ранних стадиях применяется симптоматическая терапия, корригирующая поведенческие нарушения, расстройства сна и миоклонии (амфетамины, барбитураты, антидепрессанты, бензодиазепины, другие нейролептики); в поздних - поддерживающая терапия.
Вместе с тем на современном этапе проблема разработки эффективной терапии прионных болезней считается задачей первостепенной важности, поскольку имеются прогнозы, не исключающие возможность значительной эпидемии нового варианта БКЯ в ближайшие 10 - 15 лет. Создание адекватного этиологического и патогенетического лечения больных актуально также в связи с наличием групп риска развития семейных и ятрогенных вариантов указанных заболеваний, которые могут быть выделены уже в настоящее время.
Безусловно, успехи в создании этих методов лечения зависят от существующих представлений о свойствах не только PrPSc , но и PrPC которые позволяют уже на данном этапе обсуждать некоторые из этих подходов.
Одним из наиболее перспективных путей лечения представляется предотвращение преобразования PrPC в PrPSc путем стабилизации структуры PrPC связующим активным веществом или изменением действия протеина X, который может функционировать как молекулярный шаперон. Остается определить, какой из препаратов будет более действенным: связывающий PrPC или имитирующий структуру PrPC с основными полиморфными остатками, который, возможно, предотвращает скрепи и БКЯ. Следует отметить, что средства, призванные воспрепятствовать образованию прионов, должны проникать через гематоэнцефалический барьер.
Возможным терапевтическим подходом при лечении прионных болезней может быть снижение уровня PrPC у человека без нанесения ему вреда в результате уменьшения содержания PrP мРНК с помощью олигонуклеотидов, что может отсрочить появление симптомов болезни.
Рекомендуется проводить генетический анализ прионного гена у лиц, в семьях которых были зарегистрированы больные с патологией. Более сложной является проблема пренатальной ДНК-диагностики и связанное с этим решение вопроса о прерывании беременности в случае наличия у одного из родителей наследственной прионной болезни, поскольку неполная пенетрантность некоторых из этих заболеваний делает сомнительным предсказание будущего для носителя мутантного гена.
В Европе, в том числе и в России, осуществляется ряд мероприятий по профилактике прионных инфекций. Наряду с ограничением использования лекарственных средств, приготовленных из тканей коров, прекращено производство гормонов гипофиза животного происхождения, предпочтение отдается генно-инженерным препаратам. В ряде стран введены ограничения на трансплантацию твердой мозговой оболочки. Разрабатываются запретительные положения на трансплантацию тканей, переливание крови и назначение препаратов крови от индивидуумов с деменцией.
Поскольку передача прионных болезней от человека человеку предполагает прямую инокуляцию инфекционного материала, при работе с больными в процессе инвазивных процедур, а также при контакте с их биологическими жидкостями необходимо придерживаться правил, предусмотренных при работе с больными со СПИДом. При вскрытии умерших больных применяют те же правила.
Инструменты, используемые у больных БКЯ при нейрохирургических манипуляциях, а также, по-видимому, при производстве тонзиллярной биопсии, и внутримозговые электроды должны быть уничтожены.
Еще одним путем предупреждения прионных болезней может быть разведение домашних животных, не передающих прионов. Такие примеры в природе существуют, в частности есть породы овец, генетически резистентные к скрепи. В настоящее время имеются возможности для выведения генетически резистентных пород крупного рогатого скота.
ДИАГНОСТИКА ПРИОННЫХ БОЛЕЗНЕЙ ЧЕЛОВЕКА И ИНДИКАЦИЯ ПРИОННОГО БЕЛКА
Одним из сложных вопросов является диагностика прионных заболеваний. При этом диагноз ставится клинически (при спорадической БКЯ), а морфологическая диагностика этих болезней проводится на основании исследования биопсийного или аутопсийного материалов.
Важное значение в диагностике БКЯ имеет ЭЭГ-исследование. При этом на ранних этапах болезни наблюдается замедление биоэлектрической активности.
Состав цереброспинальной жидкости при прионных болезнях обычно нормальный, воспалительная реакция в ней отсутствует. В последние годы в цереброспинальной жидкости больных БКЯ идентифицированы различные необычные белки, причем одному из них (14-3-3) придается значение при диагностике всех прионных болезней человека и животных. Следует, однако, отметить, что этот белок иногда выявляется у больных с энцефалитом и инсультом. В далеко зашедших случаях БКЯ в цереброспинальной жидкости больных регистрируется нейрональная энолаза.
В ряде исследований показано, что у больных БКЯ и у зараженных животных в сыворотке крови определяют высокие титры аутоантител к нейрофиламентам.
В настоящее время самым надежным и достоверным методом диагностики БКЯ и других прионных заболеваний является иммуноцитохимический метод выявления в биоптате отложения PrPSc . Инфекционная изоформа PrPSc откладывается в синапсах коры большого мозга и мозжечка, а также в амилоидных бляшках. Отложение PrPSc является наиболее ранним этапом в развитии БКЯ и определяется еще до развития структурных изменений в ткани мозга. Однако эта методика (иммуноцитохимическое исследование и иммуноблоттинг) находится уже за рамками чисто морфологических методов и требует специальных реактивов и оборудования.
Весьма серьезной методической стороной морфологической диагностики, будь то биопсия или аутопсия, является возможность заражения исследуемым материалом: при БКЯ опасность представляют все внутренние органы, биологические жидкости больных и особенно ткани головного и спинного мозга, а также глазные яблоки. Менее постоянно удавалось передавать заболевание с помощью введения животным цереброспинальной жидкости, ткани легких, печени, почек, селезенки и лимфатических узлов больных людей. Возникновение ятрогенных случаев БКЯ после пересадки твердой мозговой оболочки и роговицы свидетельствует о том, что прионы накапливаются не только в самом мозге, но и в связанных с ним соединительнотканных образованиях. Имеются единичные экспериментальные сообщения о том, что на определенном этапе развития БКЯ прионы могут содержаться и в крови больных.
В литературе описаны случаи заражения нейрохирурга, терапевта, стоматолога, патологоанатома и лаборантов. Следует отметить, что ткани погибших от прионных болезней остаются заразными даже после их фиксации формалином. В связи с этим работа с материалом требует особых мер предосторожности и должна выполняться специально обученным персоналом. Учитывая опасность заражения БКЯ во время хирургических вмешательств и аутопсий, важно знать, что в значительной степени опасность заражения зависит от пути проникновения инфекции. Экспериментально доказано, что наивысшей она является при интрацеребральном введении инфекционного агента, значительно уменьшается - при интраперитонеальном и становится еще более низкой при пероральном заражении. При проведении хирургических манипуляций и аутопсий должны быть предприняты меры предосторожности при работе с тканями, жидкостями и другими материалами от больных с подозрением на БКЯ во избежание возможного заражения.
ЗАКЛЮЧЕНИЕ
Изучение прионов и вызываемых ими заболеваний является новой, быстро развивающейся областью биомедицинских исследований. Проблема этих болезней, оставаясь до последнего времени экзотической в связи с их большой редкостью в человеческой популяции, в последние годы приобрела важное научно-практическое значение.
Практический интерес связан прежде всего с разразившейся эпизоотией губкообразной энцефалопатии коров в Великобритании, а также с выявлением, в основном в Великобритании, молодых людей с болезнью Крейтцфельдта-Якоба (новый вариант БКЯ) и доказательством возможности передачи этого заболевания людям в результате употребления в пищу мясопродуктов, полученных из зараженных животных. И хотя для населения России нет прямой угрозы заражения инфекционным прионным белком, на современном этапе важным является налаживание в общегосударственном масштабе работы по регистрации прионных болезней человека и животных на всей территории страны с обращением особого внимания на группы риска (работники скотоводческих и звероферм, боен, мясокомбинатов и др.).
Постоянно увеличивающийся теоретический интерес к проблеме обусловлен результатами молекулярно-биологических исследований прионов, позволивших собрать и уже в большой мере систематизировать значительный фактический материал о структуре, функции и накоплении в зараженном организме этих новых и необычных возбудителей инфекционных заболеваний человека и животных.
Именно результаты молекулярно-биологических исследований структуры прионных белков дали основание наметить новые направления в дальнейших подходах к терапии прионных болезней.
В заключение следует подчеркнуть, что все возрастающий мировой интерес к прионам и прионным болезням обусловлен в первую очередь тем, что прионы представляют собой совершенно новый класс инфекционных агентов, открытие которых без преувеличения можно сравнить по своему значению с открытием А.Левенгуком мира микроорганизмов или с открытием Д.И.Ивановским царства вирусов.
С П И С О К Л И Т Е Р А Т У Р Ы
1. Зуев В.А., Завалишин И.А., Ройхель В.М. Прионные болезни человека и животных: Руководство для врачей. – М., 1999.
2. Покровский В.И., Киселев О.И. Молекулярные основы прионных болезней. // Вестн. РАМН. – 1998. - № 10.
3. Борисов Л.В. Медицинская микробиология, вирусология, иммунология. – М., 2001.